A 71 year-old patient was referred to our attention because of a “macular dystrophy associated with optic nerve pallor” in her left eye. The patient has a history of POAG in both eyes treated with IOP-lowering drops (timolol 0.5% b.i.d, brimonidine 0.2% b.i.d, latanoprost 0.5% once a day).
She reported a fairly rapidly progressive visual acuity decline in OS over the last few months; her BCVA was 20/20 in OD and 20/200 in OS. Anterior segment examination revealed mild lens opacity in OO (which in any case still couldn’t explain her BCVA in OS) and IOP was borderline in OD and slightly higher than normal in OS (21 mmHg in OD, 24 mmHg in OS) despite medical therapy.
A complete multimodal imaging was performed in both eyes.
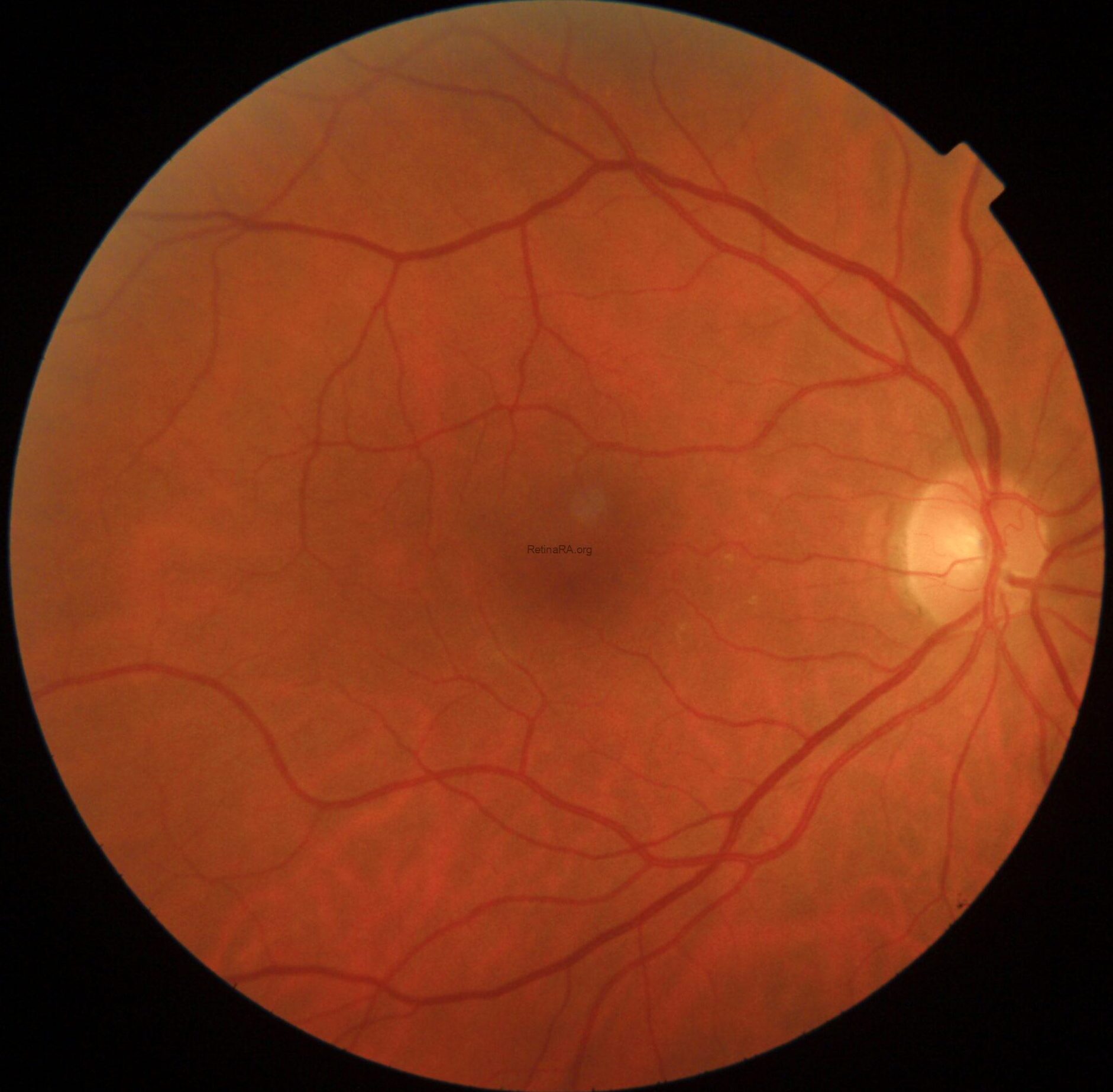
Color fundus photography in OD revealed mild RPE dystrophy in the nasal macular region and a crescent zone of temporal peripapillary chorioretinal atrophy consistent with glaucomatous beta-zone.
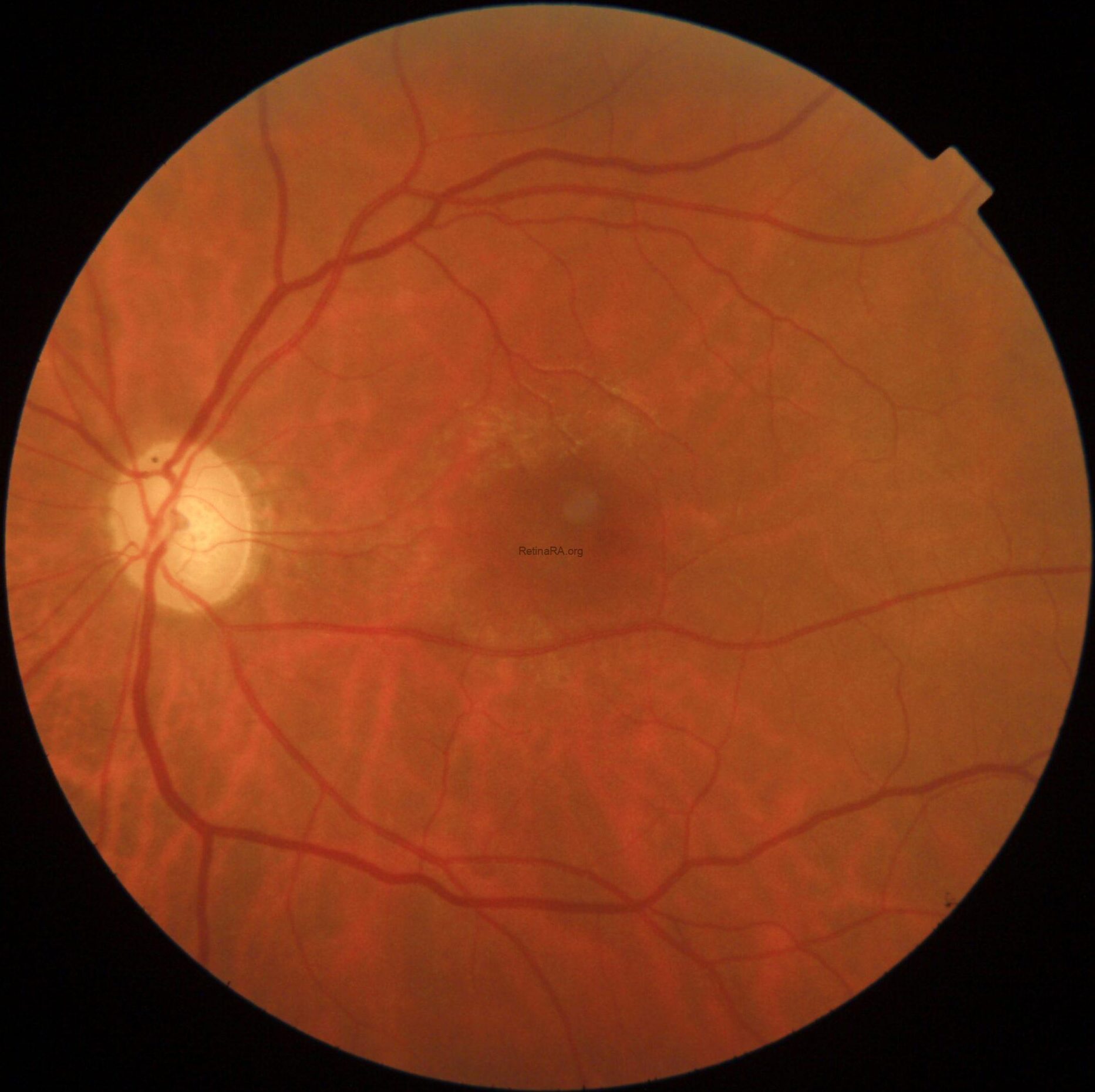
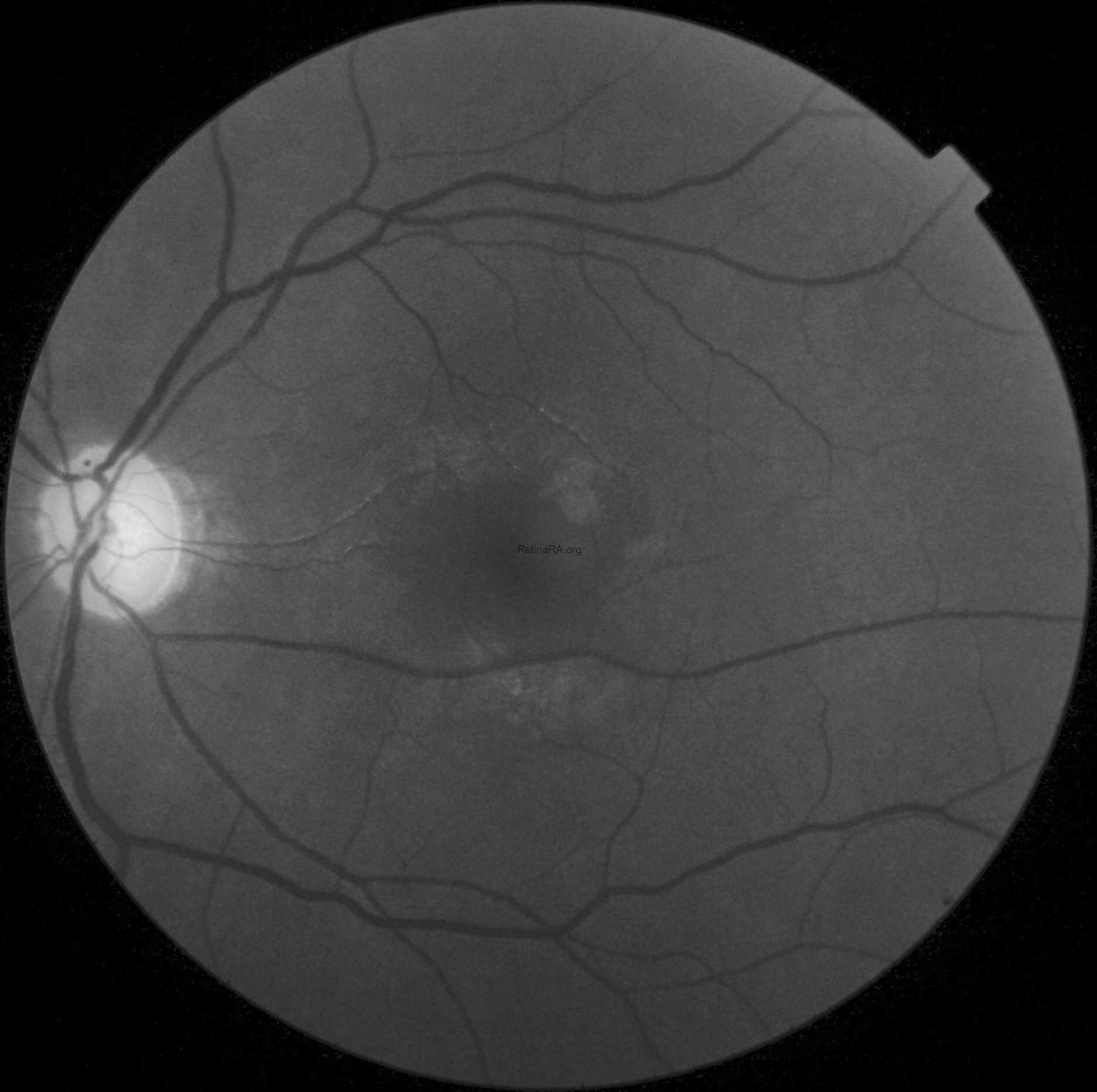
CFP in OS, however, showed marked optic nerve pallor associated with glaucomatous beta-zone temporal peripapillary atrophy, baring of circumlinear blood vessels and an extensive anomalous macular reflex, starting from the temporal edge of the optic disc and proceeding up to the perifoveal region, ultimately extending around the fovea itself.
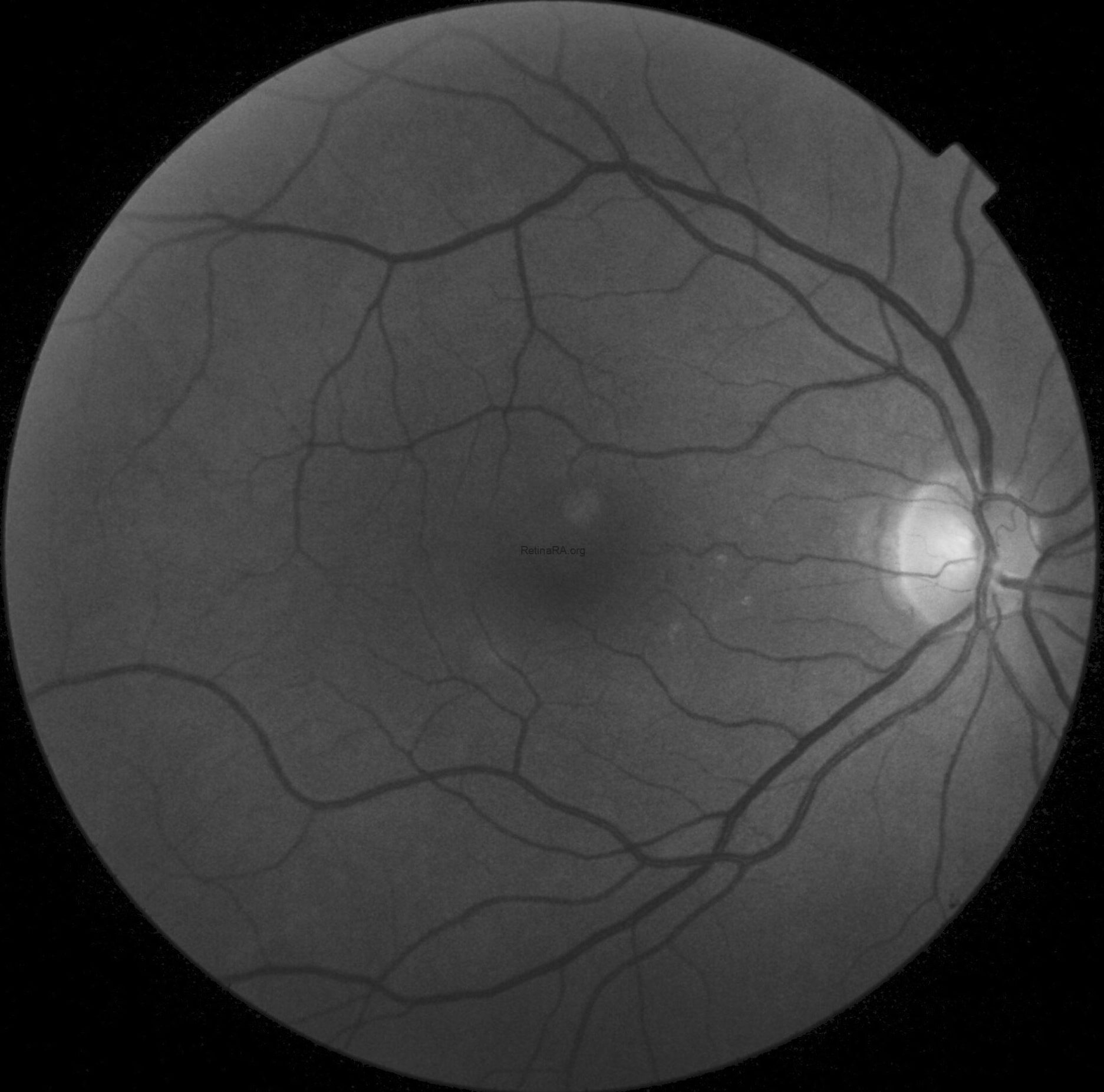
These findings are better highlighted with red-free photography, which demonstrates a full encircling of the fovea by the anomalous reflex and furthermore displays extensive RNFL defects in the left eye when compared to the right eye.
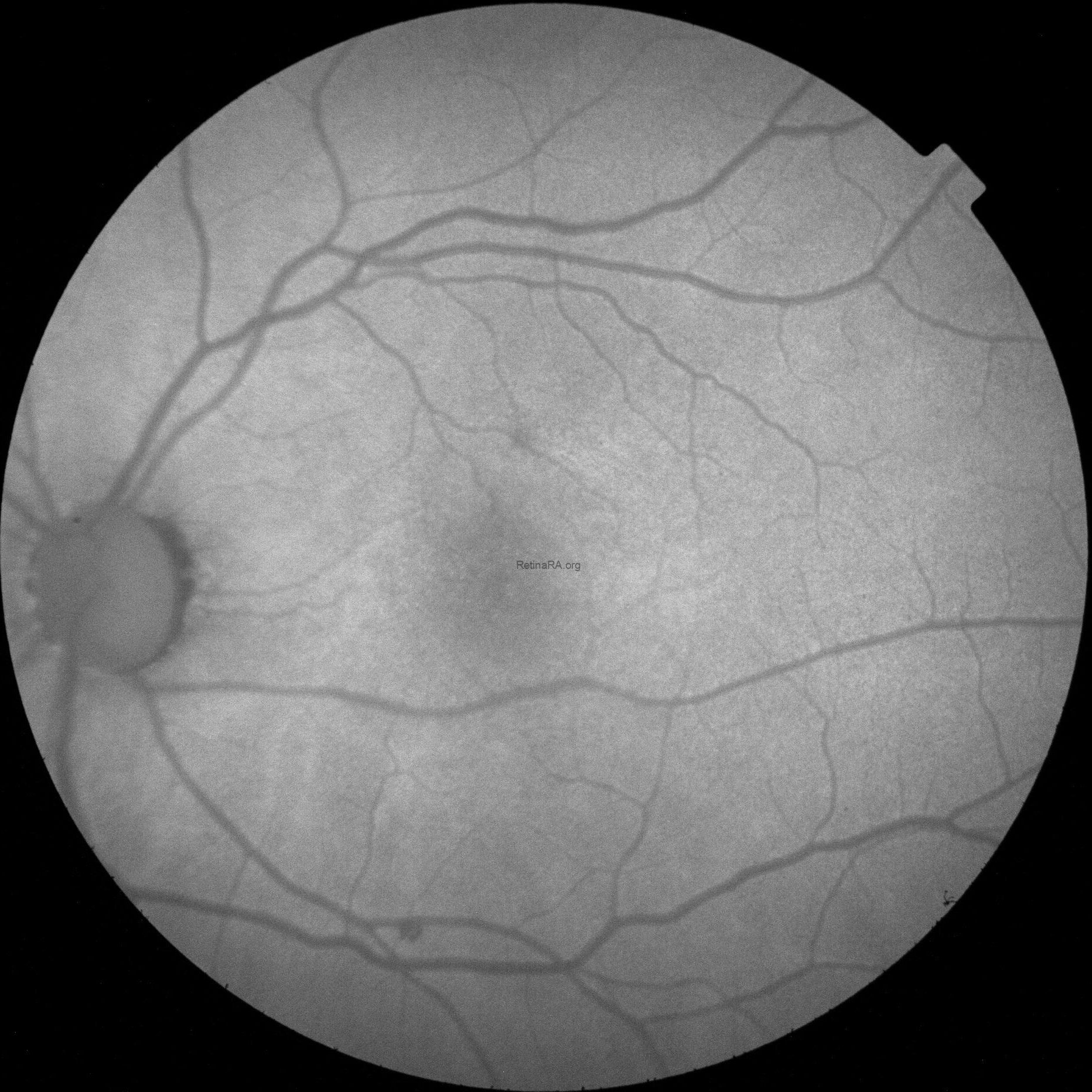
FAF in OS was normal.
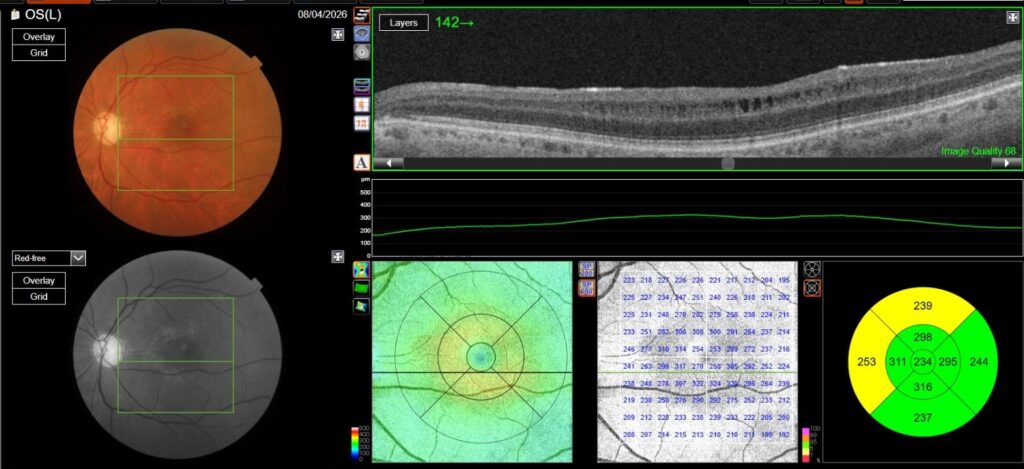
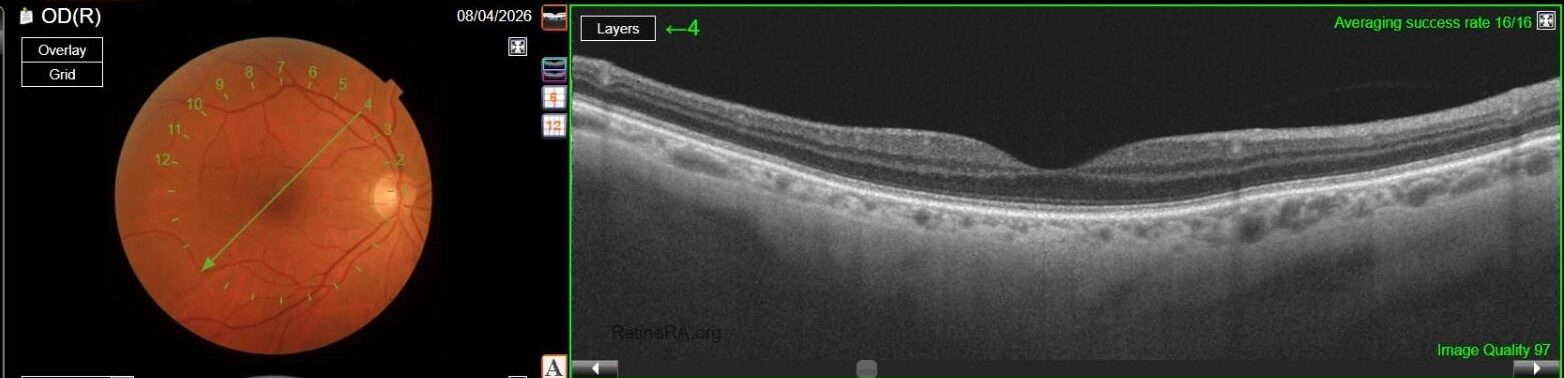
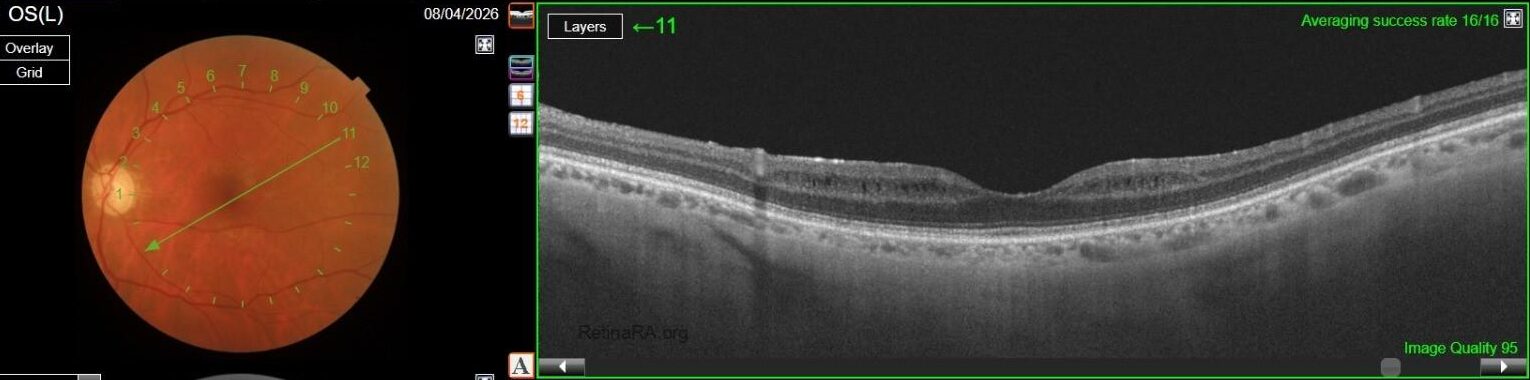
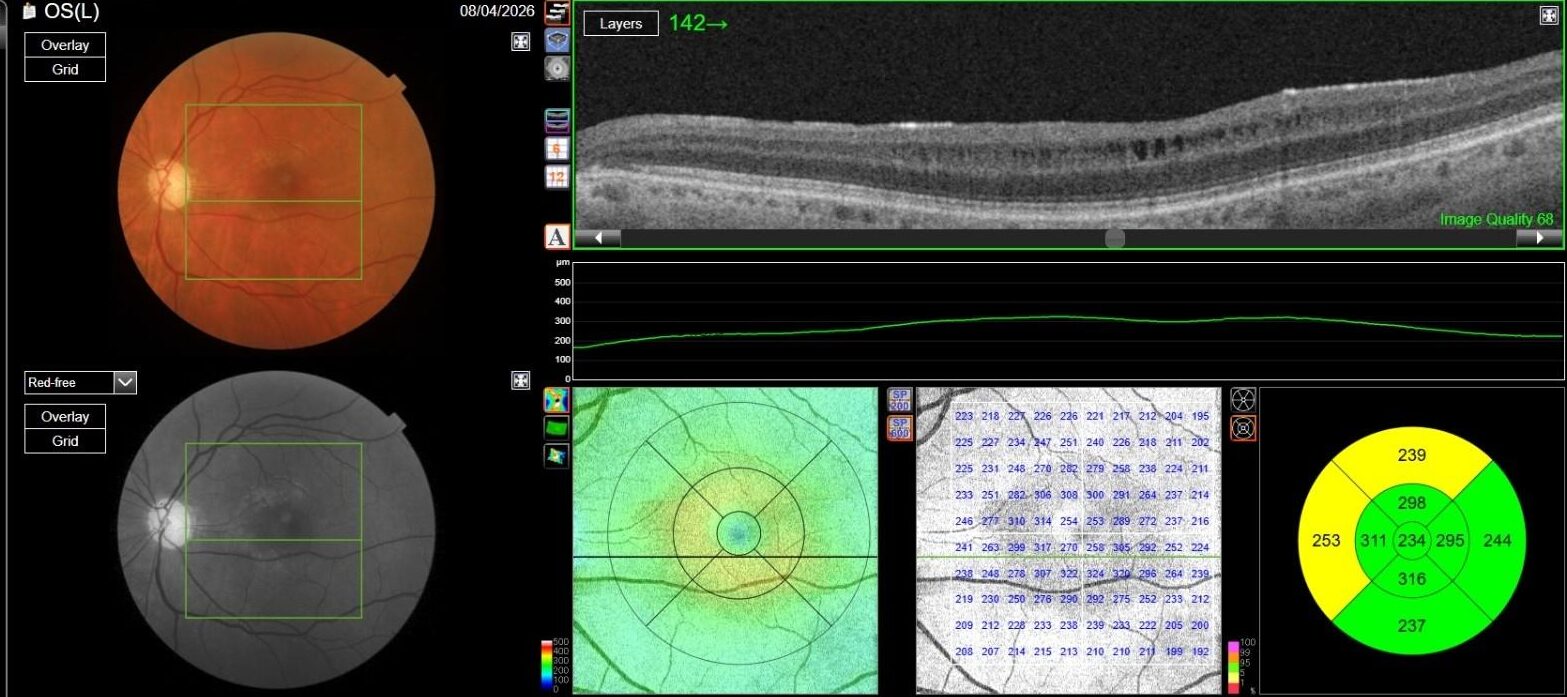
OCT in OD was normal, while in OS it showed multiple, well-defined small hyporeflective cystic spaces localized to the inner retinal layer (INL) in the perifoveal area, alongside with extreme thinning of the retinal nerve fiber layer (RNFL), whose profile and reflectivity were barely noticeable.
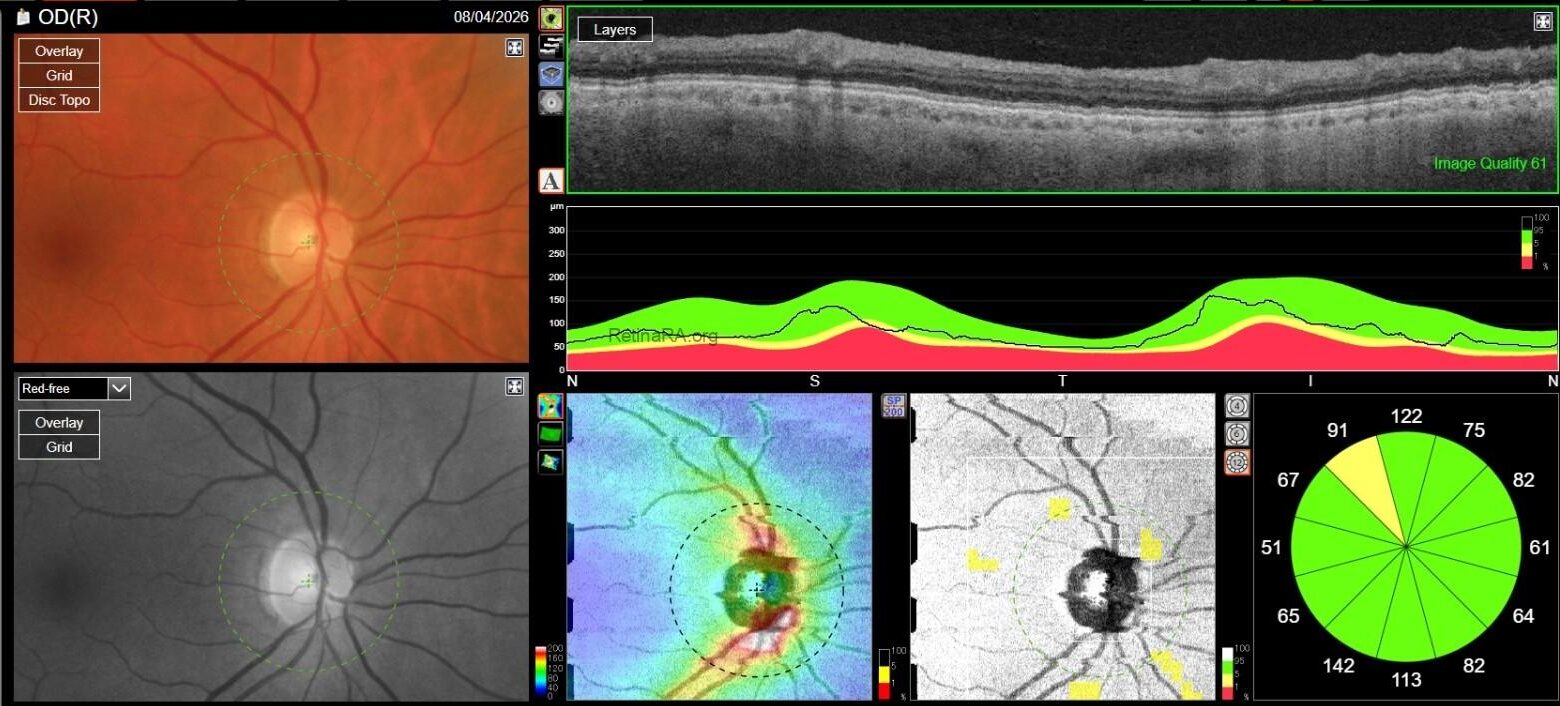
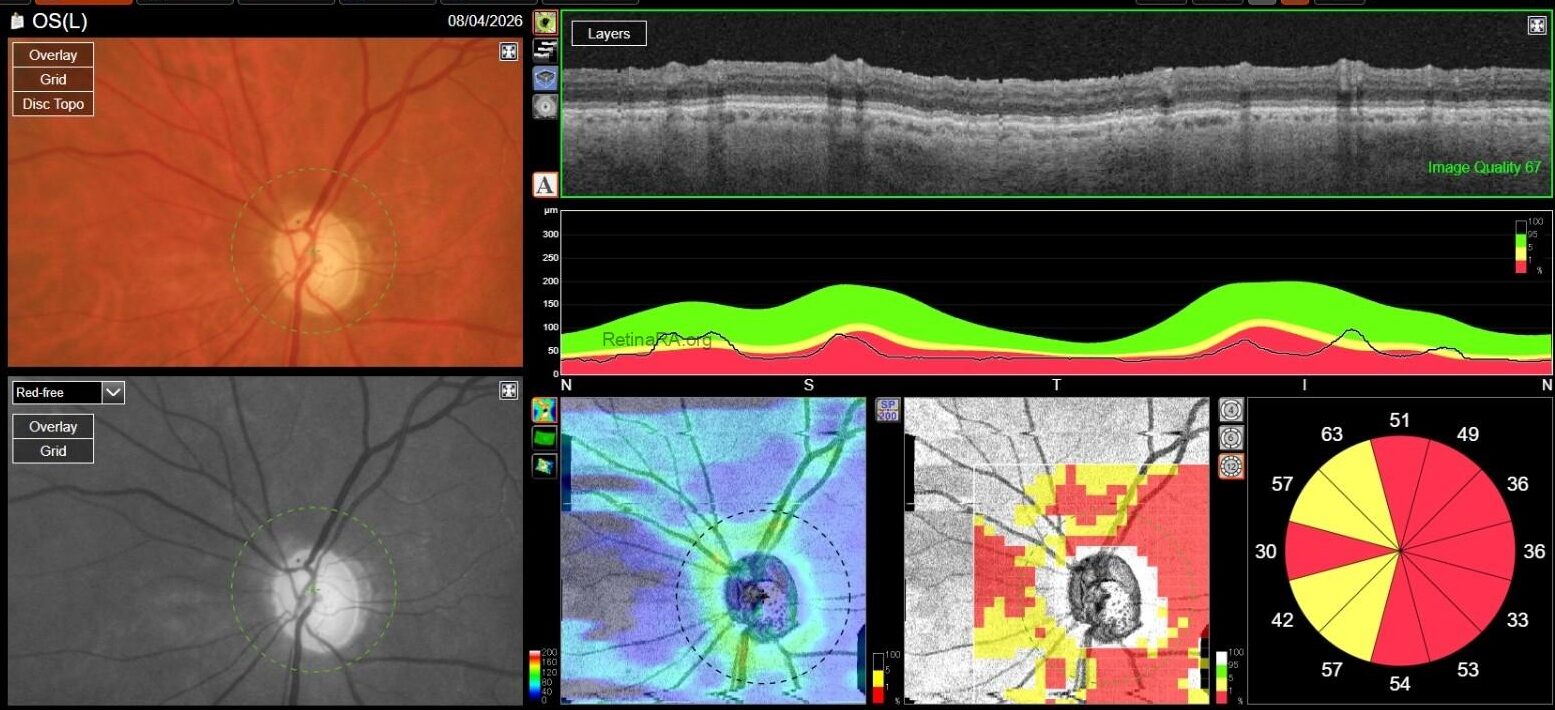
Optic Disc OCT with RNFL thickness quantification was performed as well; while in OD it revealed a minimal RNFL defect, in OS it showed extensive and severe RNFL thinning in all sectors, which was also confirmed on GCL+/GCL++ analysis by means of a 3D macula scan.
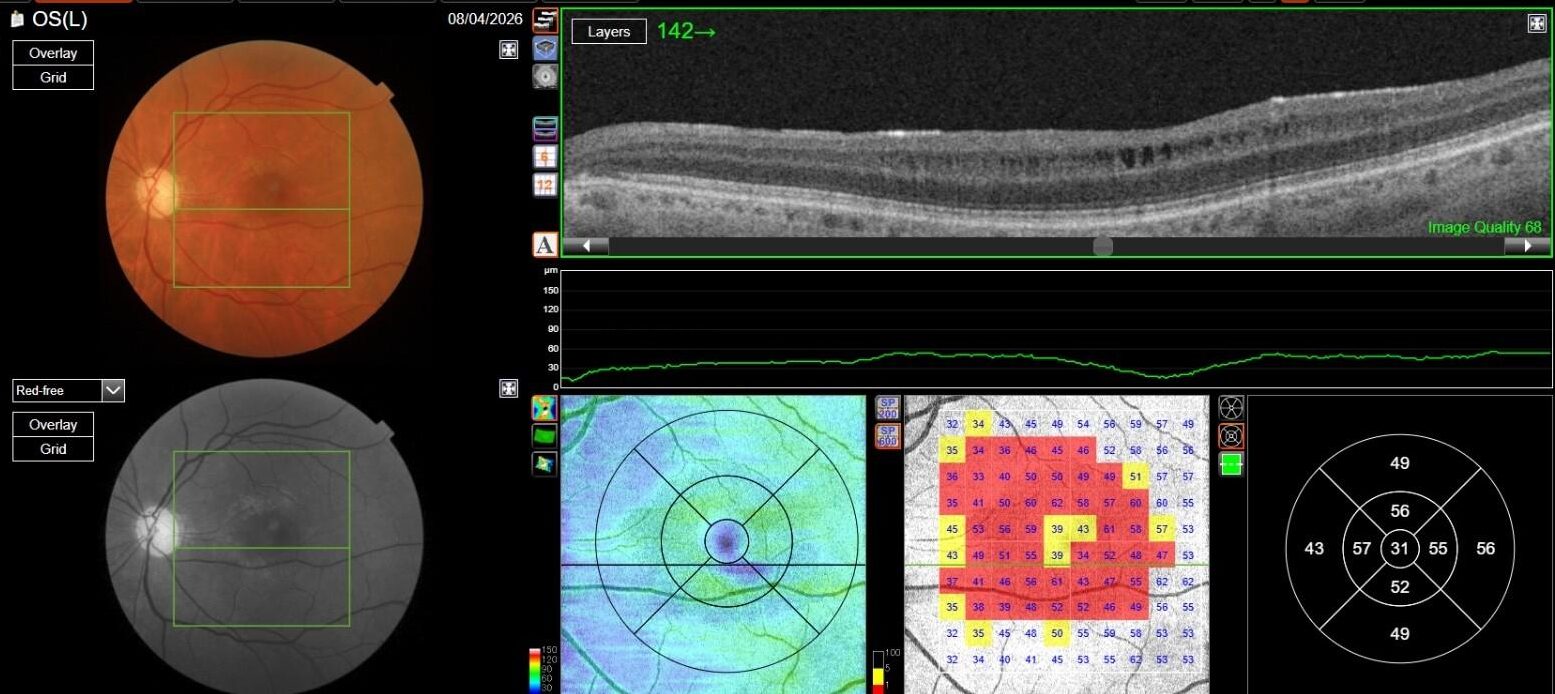
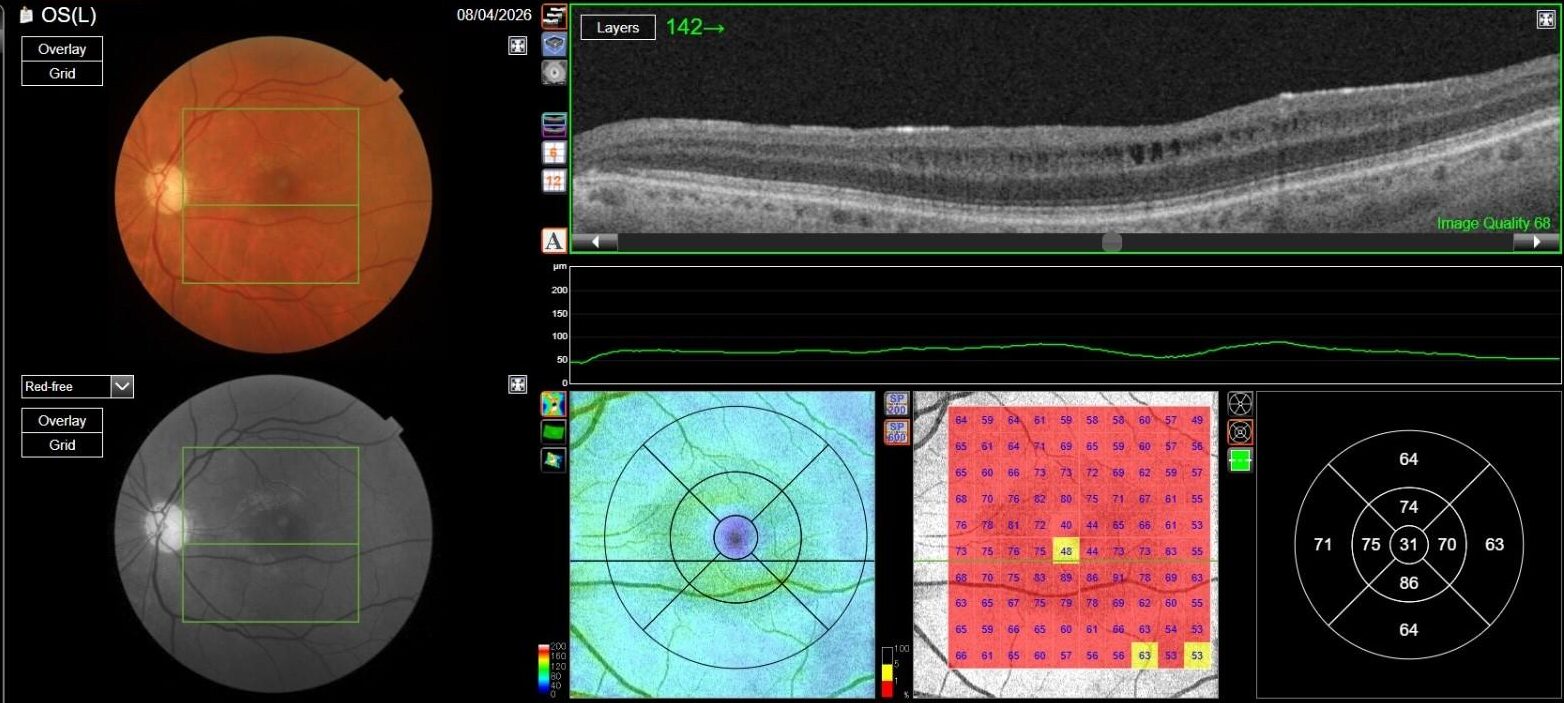
OCT-A was also performed in OS and, as expected, turned out to be negative. This also allowed an OCT en-face acquisition and subsequent segmentation at INL, which clearly showed the perifoveal distribution pattern of the INL cystic spaces previously found on b-scan OCT, with complete encompassing of the fovea and relative predominance in the inferior macular region.
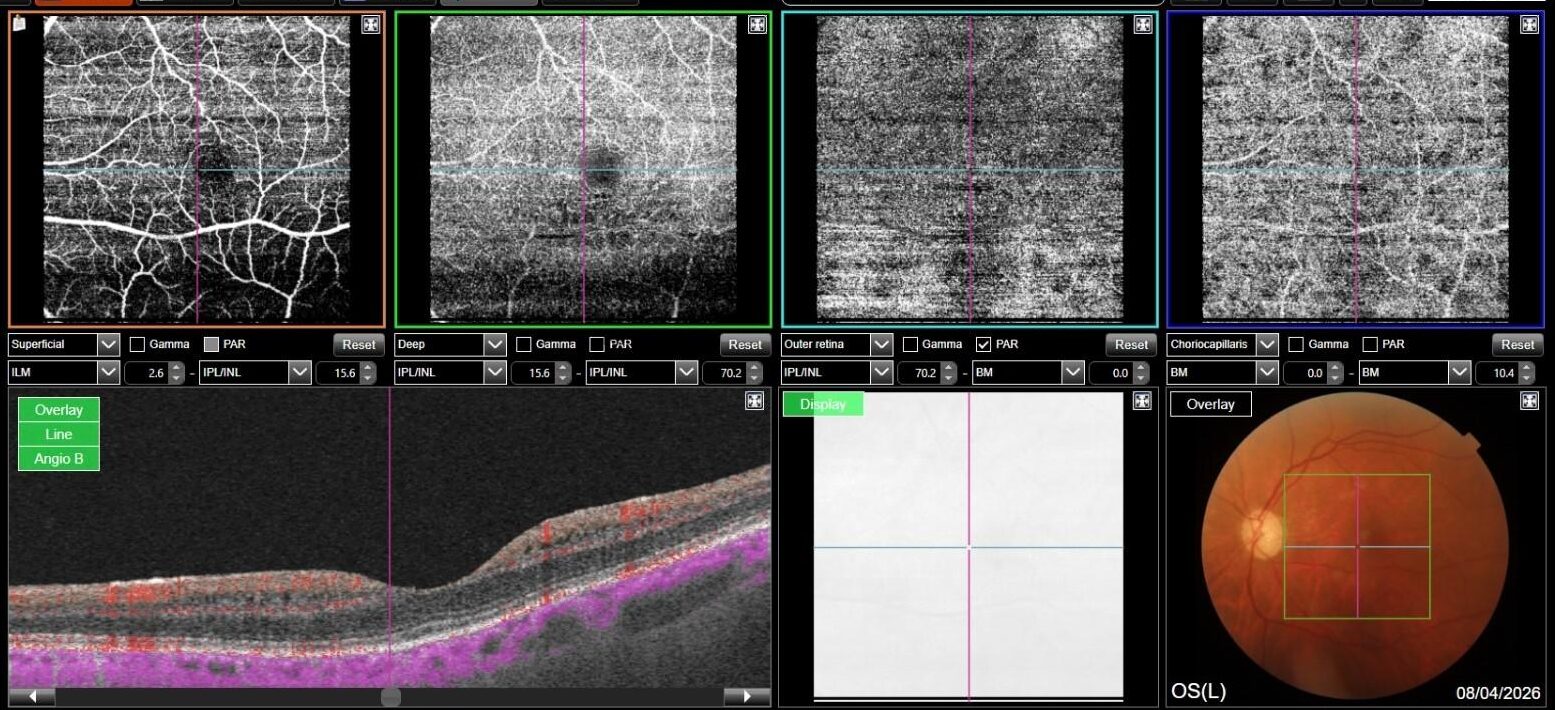
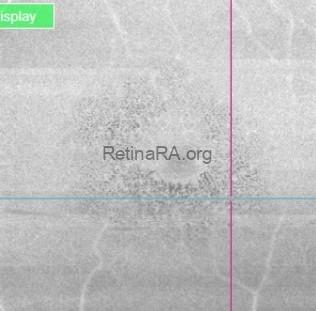
Based on patient history, clinical examination and multimodal imaging, a diagnosis of Microcystic Macular Edema (MME) secondary to Optic Neuropathy was made.
Microcystic Macular Edema (MME) is an OCT finding which was first described by Gelfand et al. in patients with Multiple Sclerosis (MS) and has since been linked to various pathological conditions classified among optic neuropathies, including Neuromyelitis Optica Spectrum Disorders (NMOSDs), hereditary optic neuropathies (such as ADOA, LHON and mitochondrial optic neuropathies), compressive optic neuropathy, traumatic optic neuropathy and glaucoma.
Several pathogenic mechanisms have been implied to play a role in MME. First of all, retrograde transsynaptic degeneration has to be taken into account. According to this mechanism, MME is caused by bipolar cell degeneration following ganglion cell death in patients with optic neuropathy, thus creating empty cystic spaces at the INL level, where bipolar cell bodies are located. This hypothesis was validated through histopathological evidence.
Another interesting model takes into consideration the retrograde degeneration of Muller cells, whose bodies are located in the INL as well. Several studies have shown that Muller cells play a pivotal role in intraretinal fluid homeostasis through acquaporin-4 (AQP-4) expression; thus, their loss could lead to a relative intraretinal fluid excess and, ultimately, to MME formation. This hypothesis could explain the relatively high prevalence of MME in the setting of NMOSDs, since these are often characterized by anti-AQP4 autoantibodies production, which could impair intraretinal water resorption by Muller cells: in particular, MME was found in 20% of patients with NMO-related optic neuritis (Gerland et al 2013), while other evidence found an even higher prevalence in similar patients, around 40% (George et al 2013).
Other evidence has also been published regarding this model in other optic neuropathies, such as MS-related optic neuritis, since MS patients are sometimes found to be positive to autoantibodies against Kir4.1, a K+ channel which is associated with acquaporin-4 and thus plays a role in intraretinal fluid resorption, which could therefore further be impaired. In NMO and MS setting, interestingly, Abegg et al. have unified these two hypothesis in a “double hit” model, according to which retrograde transsynaptic degeneration of bipolar cells is followed by Muller cell dysfunction caused by anti-AQP4 and anti-Kir4.1 autoantibodies, with subsequent MME formation in patients with optic neuritis associated to either one of these two diseases (Abegg et al 2014).
Other debated physiopathological mechanisms potentially leading to MME are vitreomacular traction (which was studied in patients with ERM and was found to be a possible concurrent cause of MME development, especially in patients already suffering from other types of optic neuropathy) and inflammation leading to glial response in the INL, which was partially refuted since MME can also be found in patients with noninflammatory optic neuropathies, such as mitochondrial optic neuropathy, traumatic optic neuropathy and glaucomatous optic neuropathy as well. Relevantly, Pott et al. reported that MME was found in around 20% of eyes with optic atrophy not related to optic neuritis over a 16-year disease period, with the most frequent optic neuropathy being traumatic optic neuropathy (Pott et al. 2016).
A common point amongst optic neuropathies related to MME development was that MME was more frequently observed in more severe disease: in both MS and NMO patients, for example, MME was associated with worse visual impairment and higher EDSS scores.
Notably, this was confirmed in glaucomatous optic neuropathy as well. Wolff et al reported that MME is associated with advanced stage glaucoma, and that MD slope was significantly different in eyes with MME compared to eyes without MME; Jung et al. found that MME was more frequent in VF progressors than in non-progressors. MME in glaucomatous eyes was predominant in the inferior hemiretina; involvement of superior hemiretina or both was still possible, but much more uncommon in the available studies.
That being said, our case allows for some interesting considerations: on one hand, MME was only found in her left eye, where the optic neuropathy was more severe, thus confirming the available literature data; on the other hand, however, en-face OCT clearly shows involvement of both inferior and superior hemiretina, although a relatively prevalent involvement of inferior hemiretina can be observed. This could either be explained by a more diffuse and severe RNFL and GC involvement, as demonstrated by both RNFL and GCC analysis, or by cohexistance of other, previously unrecognized optic neuropathy in addition to glaucomatous optic neuropathy. Lastly, no ERM was found on OCT, thus complying with evidence suggesting that vitreomacular traction may play a role in MME formation, but mainly as a concurrent cause rather than a primary pathogenetic element.
Optimization of glaucoma medical therapy was advised, since IOP was indeed higher than normal in OS; nonetheless, several elements prompted us to advice the patient to undergo further neurological assessment in order to exclude other possible optic neuropathies, such as marked asymmetry of optic neuropathy between both eyes, the self-reported fast decline in visual acuity, the finding of optic nerve pallor which was relatively more prominent than cup enlargement, the multiple associations of MME and the complete perifoveal involvement pattern.
Credit: Dr. Davide Sabbatino
Università degli Studi della Campania “Luigi Vanvitelli”
Instagram account: @ataraxia_aponia
References:
- Carlà MM, Ripa M, Crincoli E, Catania F, Rizzo S. The spectrum of microcystic macular edema: Pathogenetic insights, clinical entities, and functional prognosis. Surv Ophthalmol. 2025 Sep-Oct;70(5):982-994
- Gelfand JM, Nolan R, Schwartz DM, Graves J, Green AJ. Microcystic macular oedema in multiple sclerosis is associated with disease severity. 2012 Jun;135(Pt 6):1786-93
- Abegg M, Dysli M, Wolf S, Kowal J, Dufour P, Zinkernagel M. Microcystic macular edema: retrograde maculopathy caused by optic neuropathy. Ophthalmology. 2014 Jan;121(1):142-149
- Pott JW, de Vries-Knoppert WA, Petzold A. The prevalence of microcystic macular changes on optical coherence tomography of the macular region in optic nerve atrophy of non-neuritis origin: a prospective study. Br J Ophthalmol. 2016 Feb;100(2):216-21
- George J, Kitley J, Leite M, et al. Microcystic inner nuclear layer pathology in aquaporin-4 antibody-positive patients. Poster In: 29th Congress of the European Committee for Research and Treatment in Multiple Sclerosis (ECTRIMS) Copenhagen, Denmark, 2–5 October 2013
