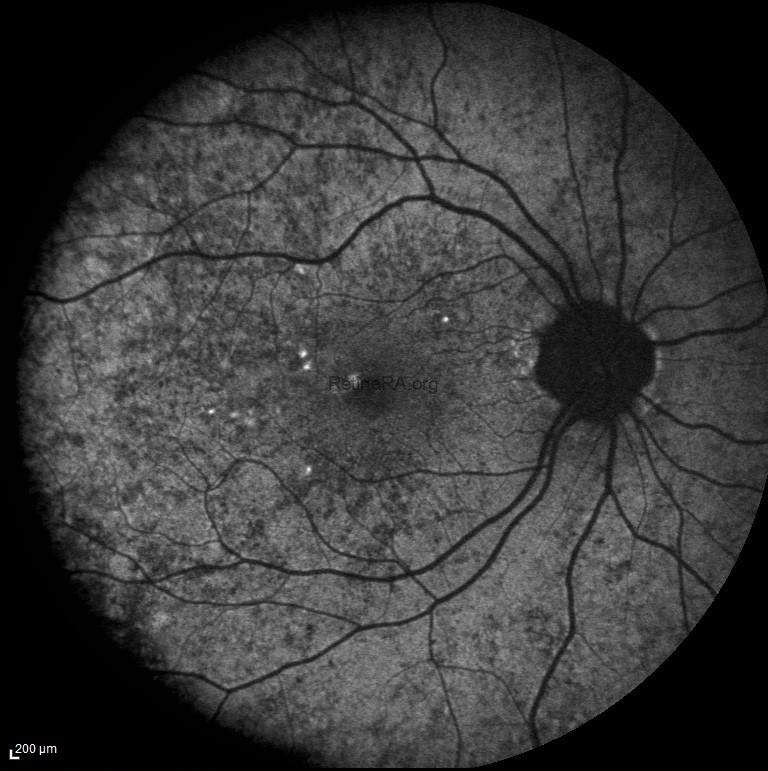
A 35-year-old female patient presented to our clinic with no main chief complaint. Her past medical history was unremarkable; however, it was noted that her father had a history of decreased vision. On ophthalmologic examination, her best-corrected visual acuity was 10/10 in both eyes, and color vision was normal bilaterally. Intraocular pressures were within normal limits. Anterior segment examination revealed no significant abnormalities. Fundus examination revealed a characteristic mosaic pattern of retinal pigment epithelium (RPE) changes, with patchy areas of hypopigmentation and hyperpigmentation, predominantly in the mid-peripheral retina. The macula appeared relatively preserved, with only subtle mottling of the RPE. The retinal vessels and optic disc were within normal limits. These findings were consistent with the phenotype observed in female carriers of choroideremia.
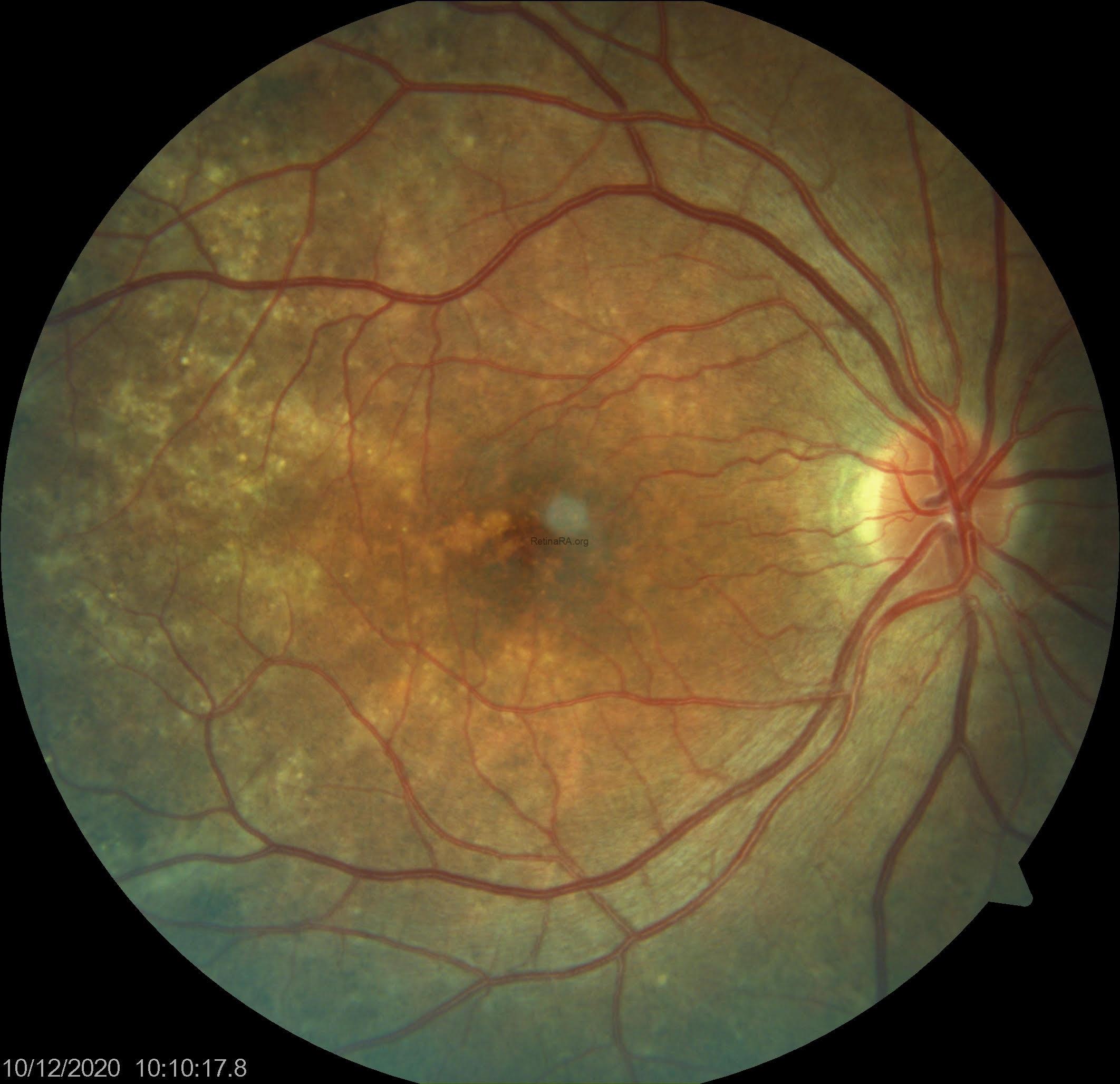

Color fundus photography reveals a distinctive mosaic pattern of retinal pigment epithelium (RPE) alterations. These images show patchy areas of hypopigmentation (corresponding to RPE atrophy or dysfunction) alternating with regions of relatively preserved RPE and pigmentation. This fundus appearance is a hallmark feature that can aid in the identification of asymptomatic or mildly symptomatic female carriers of choroideremia.
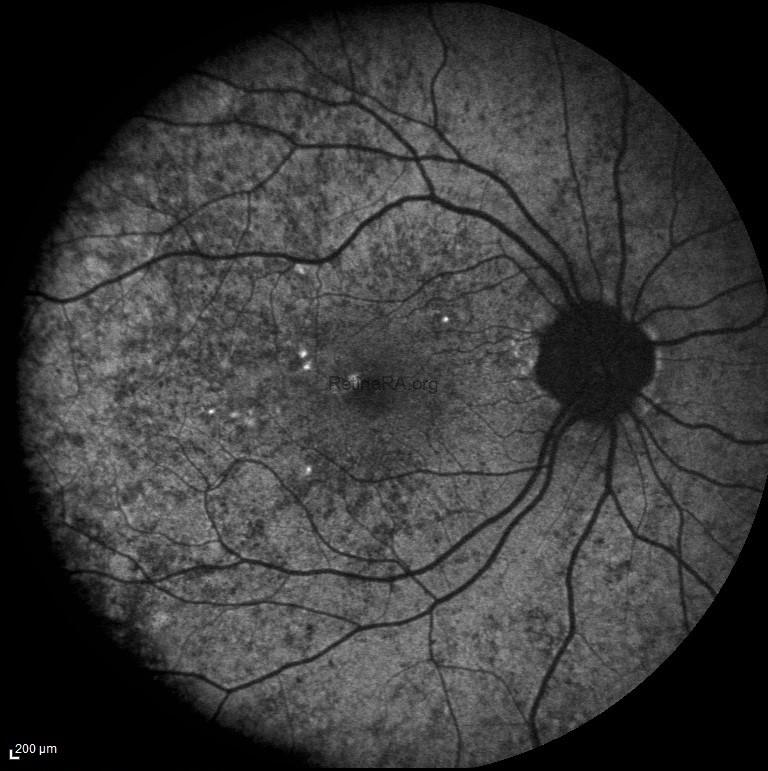

In this patient’s fundus autofluorescence (FAF) imaging demonstrates a characteristic mottled pattern of autofluorescence, reflecting the underlying mosaicism of RPE health due to X-chromosome inactivation. Areas of normal or relatively preserved RPE exhibit normal autofluorescence, while regions of RPE dysfunction or atrophy appears as zones of hypoautofluorescence.
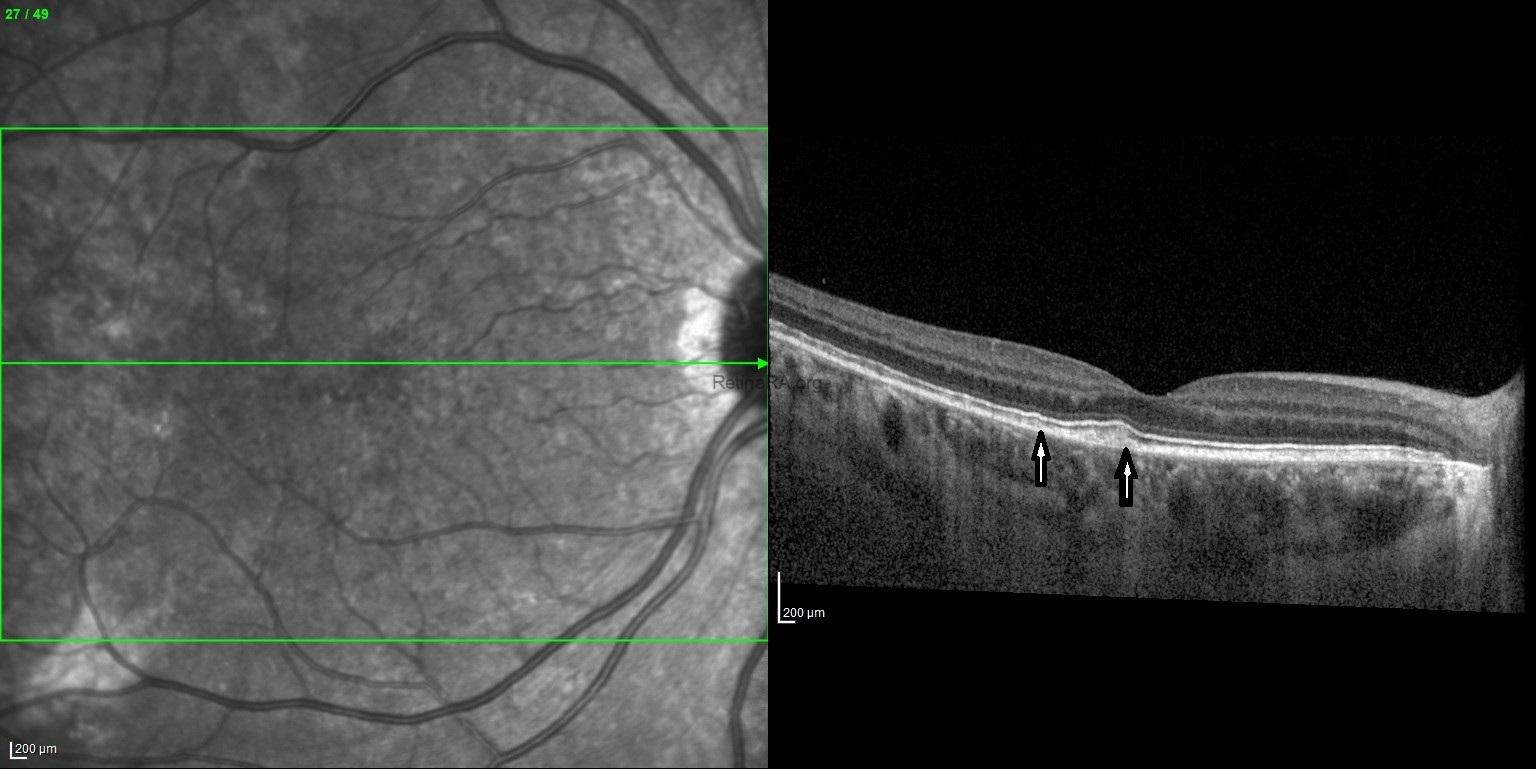

Spectral-domain optical coherence tomography (SD-OCT) reveals subtle yet characteristic structural changes that correspond to the underlying RPE mosaicism. The outer retina, particularly the ellipsoid zone (EZ) and interdigitation zone (IZ), may appear disrupted or irregular in patchy areas, especially in the mid-peripheral retina. Localized thinning or irregularity of the outer nuclear layer
(ONL) can also be observed in affected regions. The retinal pigment epithelium-Bruch’s membrane complex may show irregular reflectivity or focal attenuation in areas of RPE dysfunction. These OCT findings align with the clinical observation of a mosaic RPE pattern and can aid in the diagnosis and monitoring of disease progression in female carriers of choroideremia. Genetic analysis requested based on multimodal imaging findings proved that the patient was a choroideremia carrier.
Choroideremia is a rare X-linked recessive retinal dystrophy characterized by progressive degeneration of the choroid, retinal pigment epithelium (RPE), and photoreceptors. It predominantly affects males, while female carriers exhibit a wide range of phenotypic variability. The disease is caused by mutations in the CHM gene located on Xq21.2, which encodes Rab Escort Protein-1 (REP1). REP1 plays an essential role in intracellular vesicle trafficking by facilitating the prenylation of Rab GTPases. The absence or dysfunction of REP1 disrupts normal cellular transport mechanisms, ultimately compromising the viability of photoreceptors, RPE cells, and the choroidal vasculature.
As an X-linked disorder, choroideremia is typically expressed in hemizygous males who inherit the mutant CHM gene from their carrier mothers. Female carriers possess one normal and one mutant X chromosome. Due to random X-chromosome inactivation (lyonization), carriers display a mosaic pattern of retinal involvement, leading to variable phenotypic expression. In males, the disease generally presents during the first or second decade of life with symptoms of nyctalopia (night blindness), followed by progressive constriction of peripheral visual fields. Central vision tends to remain relatively intact until the fifth or sixth decade, although significant visual impairment eventually occurs as the disease advances. The degeneration of the chorioretinal tissues begins in the mid-peripheral retina and gradually extends both centrally and peripherally. In the late stages, only a small central island of vision remains, which is ultimately lost in many patients.
In conclusion, choroideremia represents a unique form of retinal dystrophy marked by progressive degeneration of the choroid, RPE, and outer retina. The clinical spectrum ranges from profound, progressive visual impairment in affected males to variable, often subclinical changes in female carriers. Multimodal imaging — including color fundus photography, OCT, FAF, FFA, and OCTA — provides a comprehensive assessment of disease status, aids in early diagnosis, and facilitates monitoring of disease progression and therapeutic response. With ongoing research into gene therapy and other treatment modalities, these imaging tools will play a pivotal role in the future management of choroideremia.
Credit: M. Giray Ersoz, MD, FEBO, Retina Specialist
Memorial Bahçelievler Hospital, Department of Ophthalmology, Istanbul, Turkey
Arel University School of Medicine, Department of Ophthalmology, Istanbul, Turkey
Instagram accounts: @retina.review and @retina.dr.girayersoz
Website: www.girayersoz.com.tr
and Sepideh Lotfi, MD
Biruni University School of Medicine, Department of Ophthalmology, Istanbul, Turkey
Instagram accounts: @sepidls
