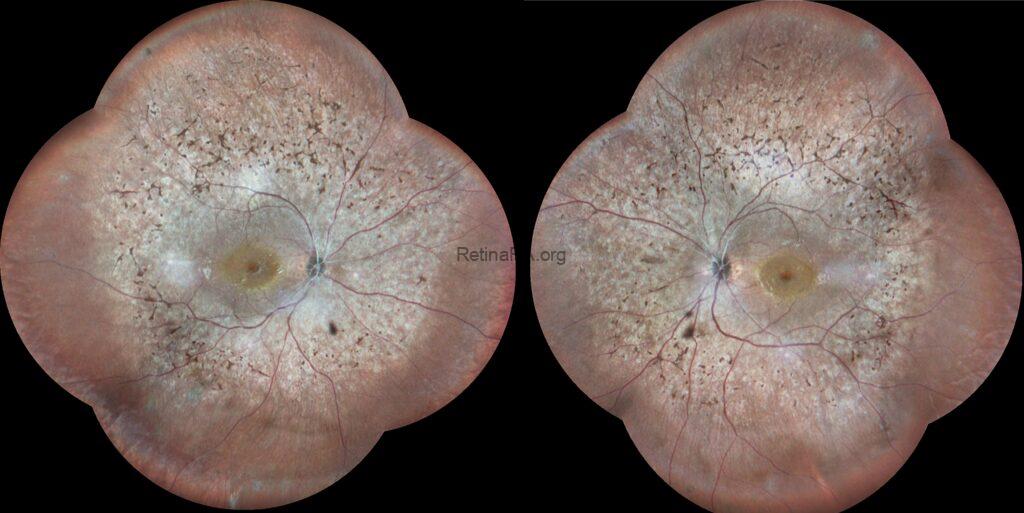
This was a 30-year-old female suffering from decreased vision and impairment of night vision and loss of peripheral vision. The BCVAs were 20/200 for both eyes, and IOPs were within normal limits. Biomicroscopic evaluation revealed a mild posterior subcapsular cataract.
Wide-field multicolor images demonstrated midperipheral retinal hyperpigmentation in the form of bone-spicule and pigment clumpings in addition to arteriolar attenuation in both eyes.


Optical coherence tomography images of both eyes showed diffuse atrophy of the outer retinal layers, including the ellipsoid zone, external limiting membrane, and RPE, except for the fovea. Irregular and disorganized outer retinal layers were also detected in the foveal regions.


Fundus autofluorescence exhibited diffuse, patchy hypoautofluorescence in the mid-periphery with abnormal perimacular hyperautofluorescence.


Retinitis pigmentosa (RP) is a group of genetic eye disorders characterized by the progressive degeneration of photoreceptor cells in the retina, leading to vision loss. Usually, RP begins with damage and loss of rod cells, leading to nyctalopia and defective dark adaptation, which are the common initial concerns for patients. It eventually progresses to involve cone photoreceptors, resulting in central and colour vision loss. Fundus features include variable amounts of bone spicule pigmentation, retinal pigment epithelium atrophy or depigmentation, retinal arteriolar attenuation, and optic disc pallor. A significant proportion of RP patients may also develop cataracts and/or cystoid macular edema.
Credit: Kemal Tekin, M.D., from Ulucanlar Eye Training and Research Hospital
Instagram accounts: @retina.academy and @dr.kemaltekin