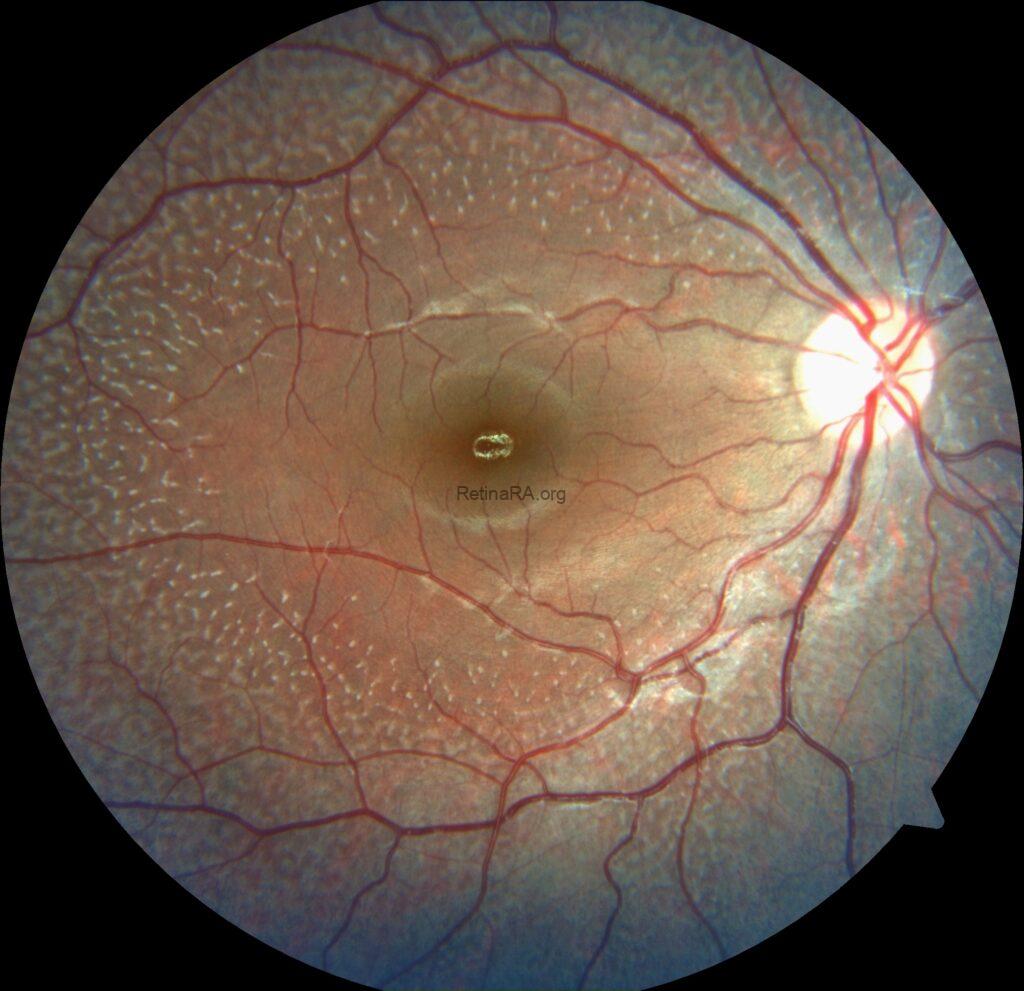
An 18-year-old female complaining of night visual difficulties was referred to our clinic. Her visual acuity was 20/20 in both eyes. She was diagnosed with fundus albipunctatus based on her fundus appearance and autofluorescence image, as well as optical coherence tomography findings. The diagnosis was then confirmed through genetic testing.
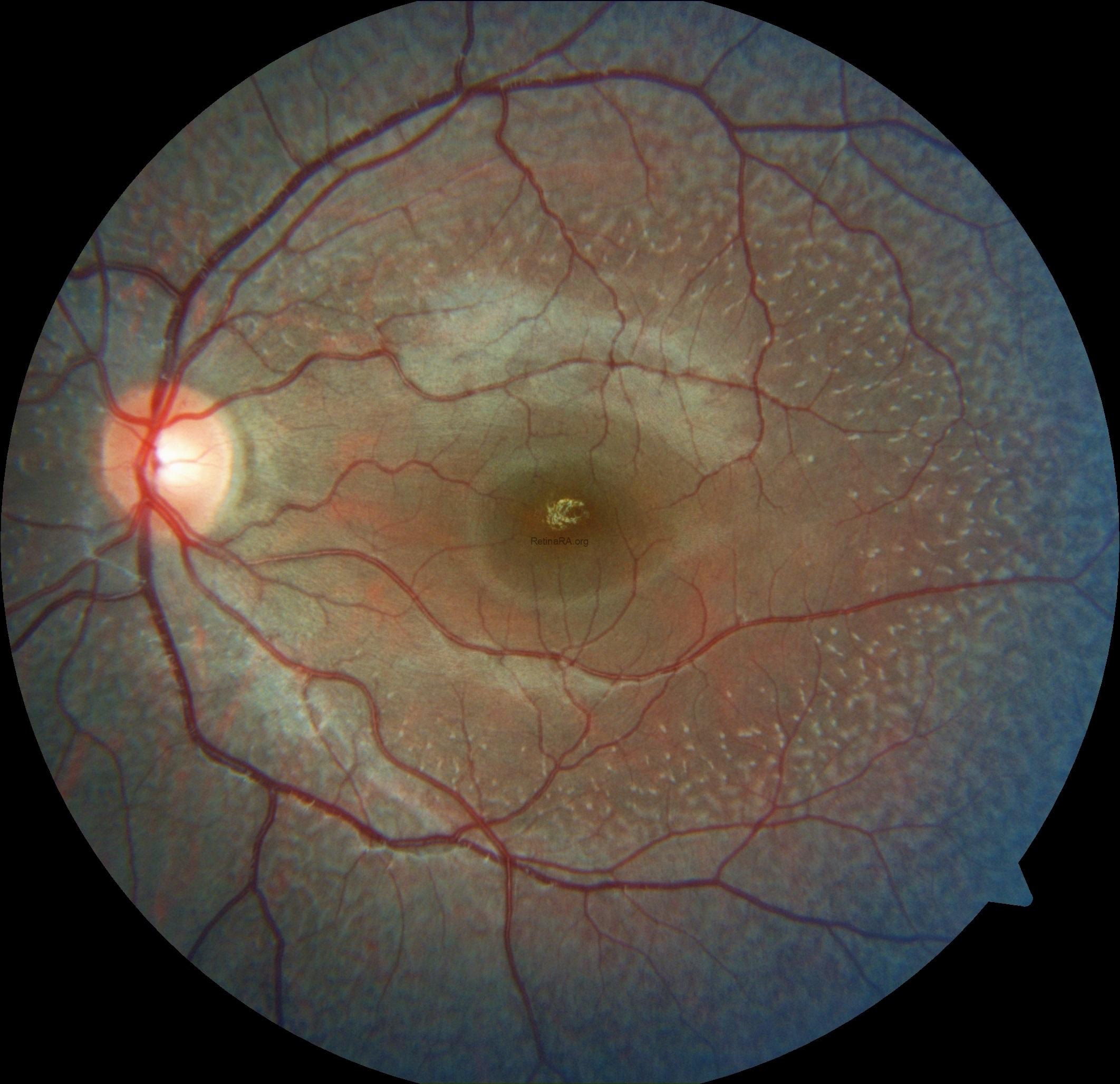
Fundus albipunctatus is an uncommon inherited retinal disorder classified within the spectrum of congenital stationary night blindness. It follows an autosomal recessive inheritance pattern and is clinically defined by numerous small white to yellow-white punctate lesions distributed throughout the posterior pole and extending into the mid-peripheral retina, typically sparing the fovea, although macular involvement has been reported in some cases. The condition is most frequently associated with pathogenic variants in the RDH5 gene, which encodes 11-cis retinol dehydrogenase, a key enzyme in the visual (retinoid) cycle responsible for the regeneration of visual pigment.
Ophthalmoscopic examination reveals scattered, discrete yellowish-white dots at the level of the retinal pigment epithelium, predominantly outside the macula. While these lesions are generally stable over time, partial regression has been documented in long-term follow-up. Histopathologically and functionally, these deposits are thought to represent accumulated retinoid intermediates, extending from the RPE–Bruch membrane complex into the outer retinal layers, including the outer nuclear layer.


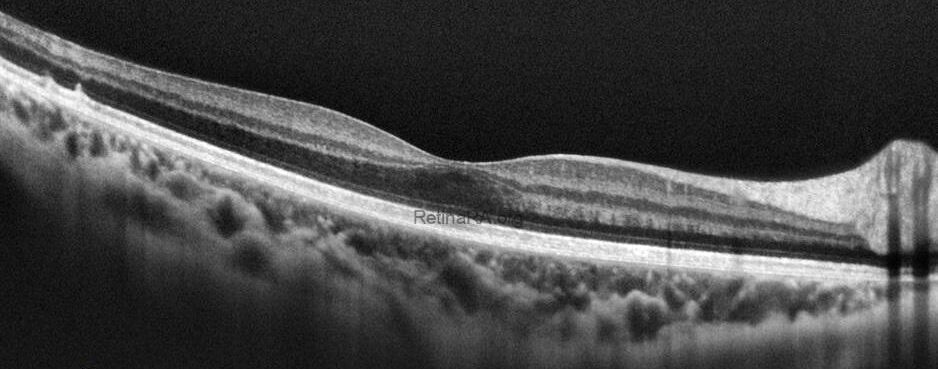
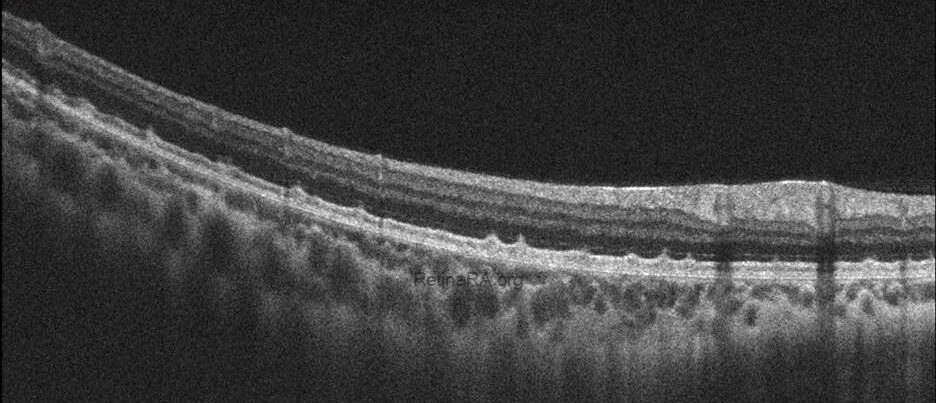
Optical coherence tomography (OCT) provides valuable structural information in fundus albipunctatus. OCT imaging typically demonstrates focal hyperreflective material originating from the RPE and projecting into the outer retinal layers, correlating with the punctate lesions seen on fundus examination. On fundus autofluorescence imaging, a diffusely reduced background autofluorescence is commonly observed, reflecting impaired retinoid metabolism and RPE dysfunction related to disruption of the visual cycle.

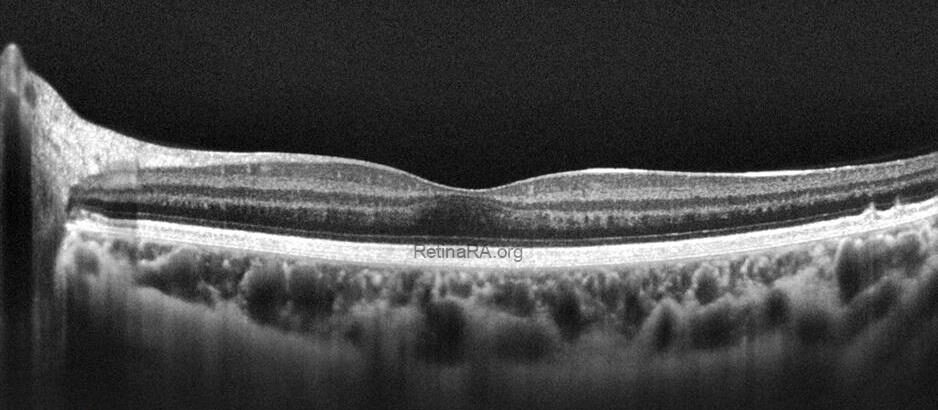
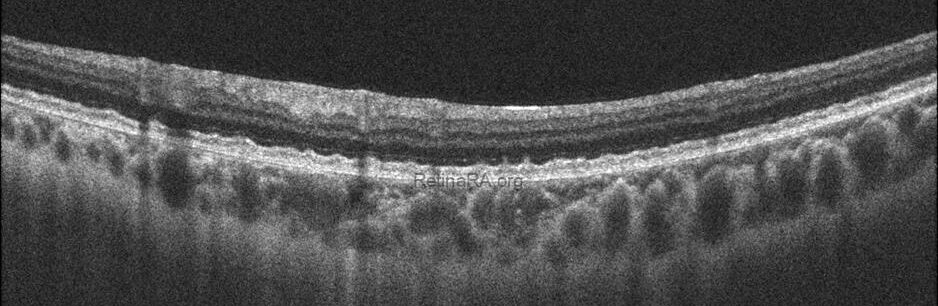
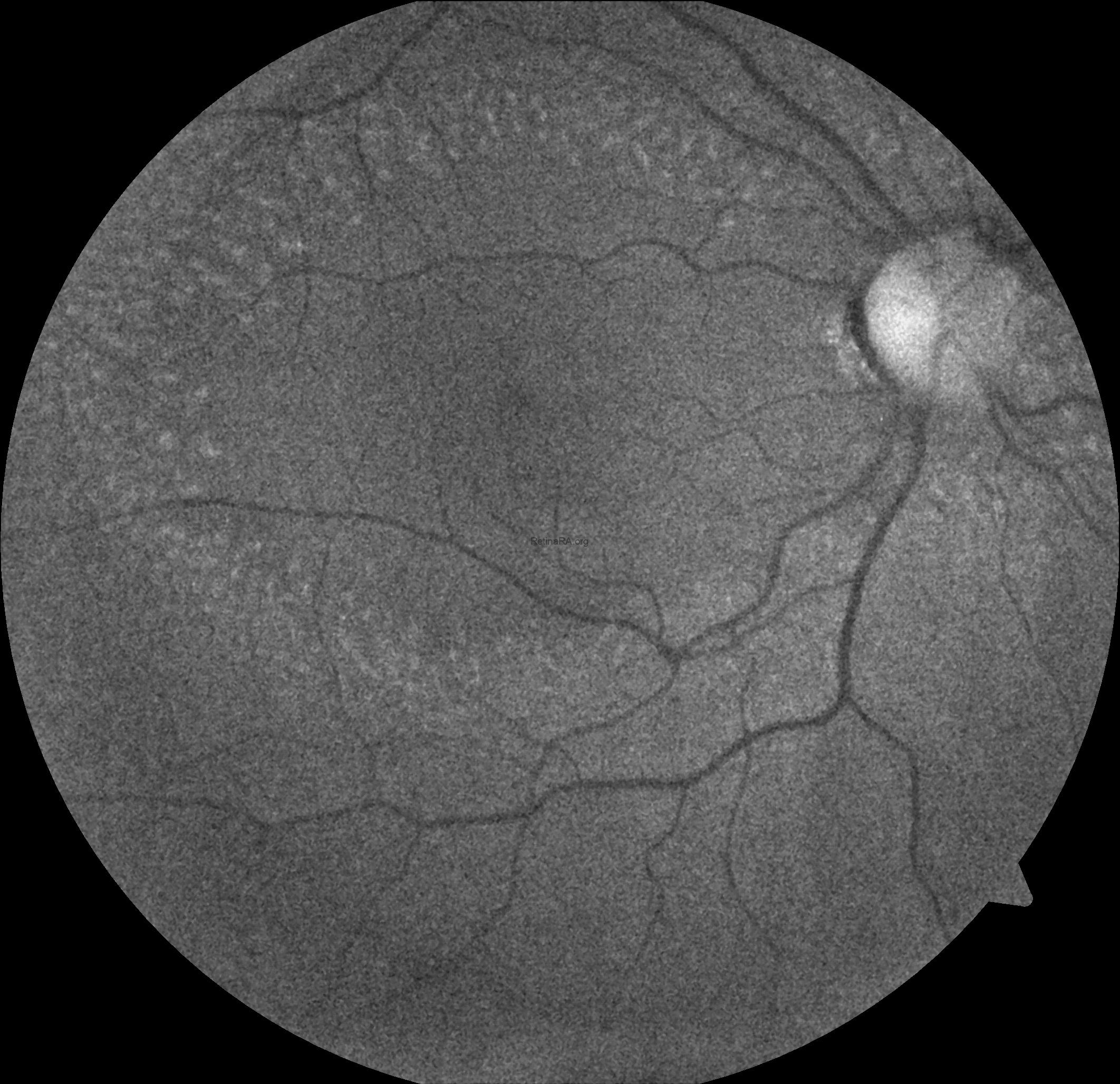
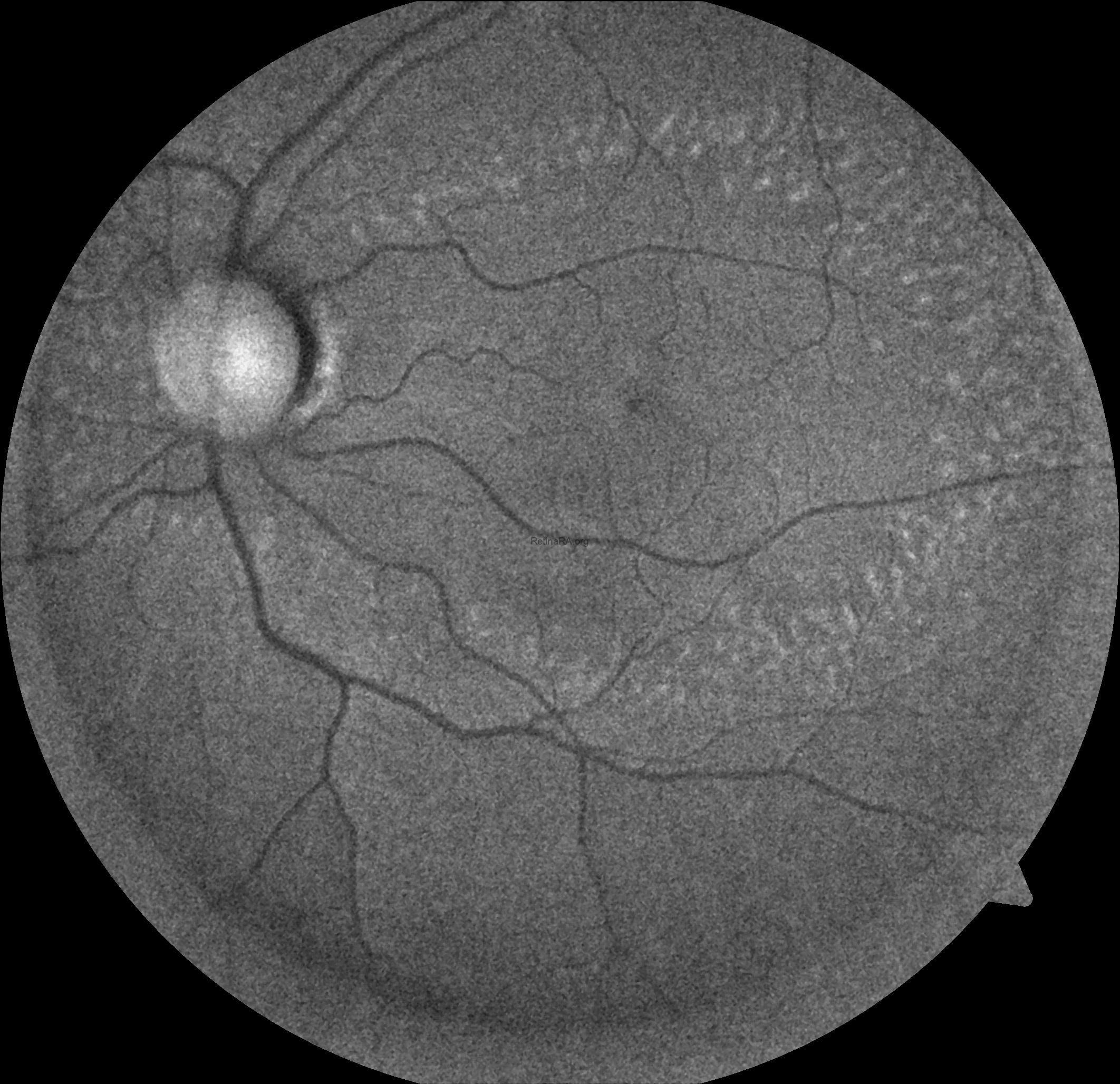
Functionally, patients exhibit a profoundly delayed dark adaptation, affecting both rod and cone systems. Dark adaptation curves show a markedly prolonged recovery phase, particularly for rod-mediated vision. Full-field electroretinography mirrors this functional delay, with subnormal responses after standard dark adaptation periods that gradually normalize following extended dark adaptation, which may require several hours and varies considerably among individuals. Once adequate dark adaptation is achieved, ERG amplitudes and psychophysical thresholds can return to near-normal levels. Similarly, electrooculography findings parallel ERG responses, as a normal light peak is typically only obtained after prolonged dark adaptation sufficient to restore visual cycle function.
Credit: M. Giray Ersoz, MD, FEBO
Memorial Hospital, Department of Ophthalmology, Istanbul, Turkey
Instagram accounts: @retina.review and @retina.dr.girayersoz