A 34-year-old female patient presented with a 7–8-year history of progressively worsening vision and increasing light sensitivity in both eyes. Visual acuity was 1/10 in both eyes, improving to 3/10 with sunglasses. Color vision was impaired, with 7/24 Ishihara plates correctly identified. An external center had previously performed an ocular tumor work-up, and cranial–orbital MRI revealed no pathology. Anterior segment examination was unremarkable, intraocular pressure was 16 mmHg bilaterally, and no vitritis was detected. Family history was noncontributory.




On color fundus photography, a lesion measuring approximately one-half of the optic disc diameter is observed in the superotemporal quadrant of the optic disc, consistent with an optic disc melanocytoma. It appears that the ocular tumor work-up was performed to evaluate this finding; however, the lesion is likely incidental. Apart from the melanocytoma, no additional remarkable abnormalities are noted in either eye.


Wide-field fundus imaging also shows no additional remarkable abnormalities.




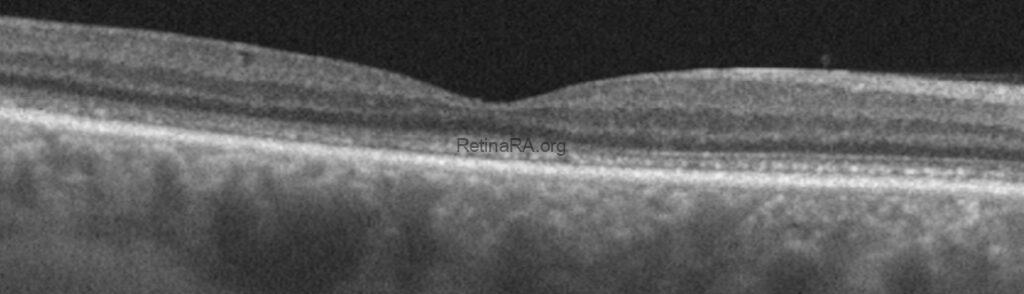
On initial inspection, OCT reveals no significant pathology in either eye.


However, upon closer examination, there is noticeable thinning of the outer nuclear layer, a thickened and disrupted appearance of the ellipsoid zone, and the interdigitation zone is not discernible.


On macular thickness maps, a noticeable thinning is evident in both eyes, particularly involving the foveal center and the nasal macular region.


In the right eye, fluorescein angiography shows no remarkable findings in either the early or late phases.


In the left eye as well, fluorescein angiography demonstrates no pathological findings in early and late phases.


On 24-2 visual field testing, central visual field defects were observed in both eyes, whereas the more peripheral test locations remained within normal limits.


Multifocal ERG demonstrated reduced central amplitudes.
Pattern ERG revealed reduced P50 amplitudes, with the decrease more pronounced in the left eye than in the right. A very subtle cone amplitude reduction was also detected on flash ERG.
In light of these findings, and particularly based on the OCT appearance, occult macular dystrophy was suspected, with a pathogenic variant in the RP1L1 gene considered the most likely underlying cause. Contrary to our initial expectation, genetic testing did not reveal a pathogenic variant in the RP1L1 gene. Instead, a heterozygous CRX gene defect was identified, which, although uncommon, has been reported to cause an occult macular dystrophy–like phenotype or an occult macular dysfunction syndrome.
Occult macular dystrophy (OMD) is an inherited macular disorder characterized by progressive central visual loss despite a clinically normal fundus and preserved full-field electroretinography. The condition is classically associated with pathogenic variants in the RP1L1 gene, which disrupt photoreceptor integrity—particularly the ellipsoid and interdigitation zones—leading to subtle but progressive outer retinal dysfunction. Patients often demonstrate decreased visual acuity, photophobia, and impaired color discrimination, while structural OCT reveals outer nuclear layer thinning and irregularity or loss of photoreceptor bands that may be overlooked on routine examination. Although RP1L1 mutations are most commonly implicated, other genes involved in photoreceptor transcription and maintenance, such as CRX, have occasionally been reported to produce an OMD-like phenotype, broadening the genotypic and phenotypic spectrum of this underrecognized macular disorder.
Credit: M. Giray Ersoz, MD, FEBO
Memorial Bahçelievler Hospital, Istanbul, Turkey
Instagram accounts: @retina.review and @retina.dr.girayersoz
and Sepideh Lotfi, MD
Biruni University School of Medicine, Department of Ophthalmology, Istanbul, Turkey
Instagram accounts: @sepidls