
A 20-year-old female presented to our clinic with complaints of progressive visual decline. Her medical history included sensorineural hearing loss, chronic renal failure, and polydactyly surgery during childhood. There was a history of parental consanguinity. Best-corrected visual acuity was counting fingers at 2 meters in both eyes. Anterior segment examination revealed bilateral posterior subcapsular cataracts. Intraocular pressure was within normal limits. Fundus examination revealed attenuated retinal vessels, waxy pallor of the optic disc, and bone spicule-like pigmentary deposits in the mid-periphery.
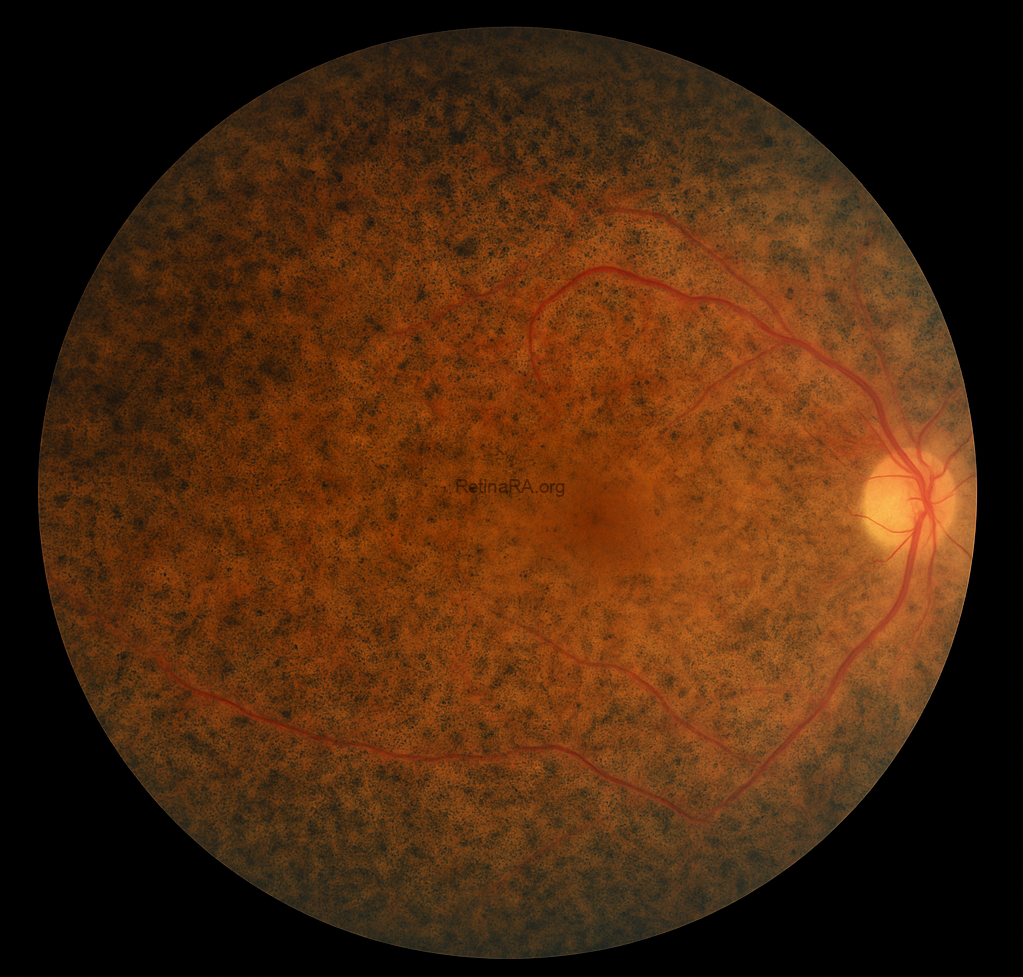
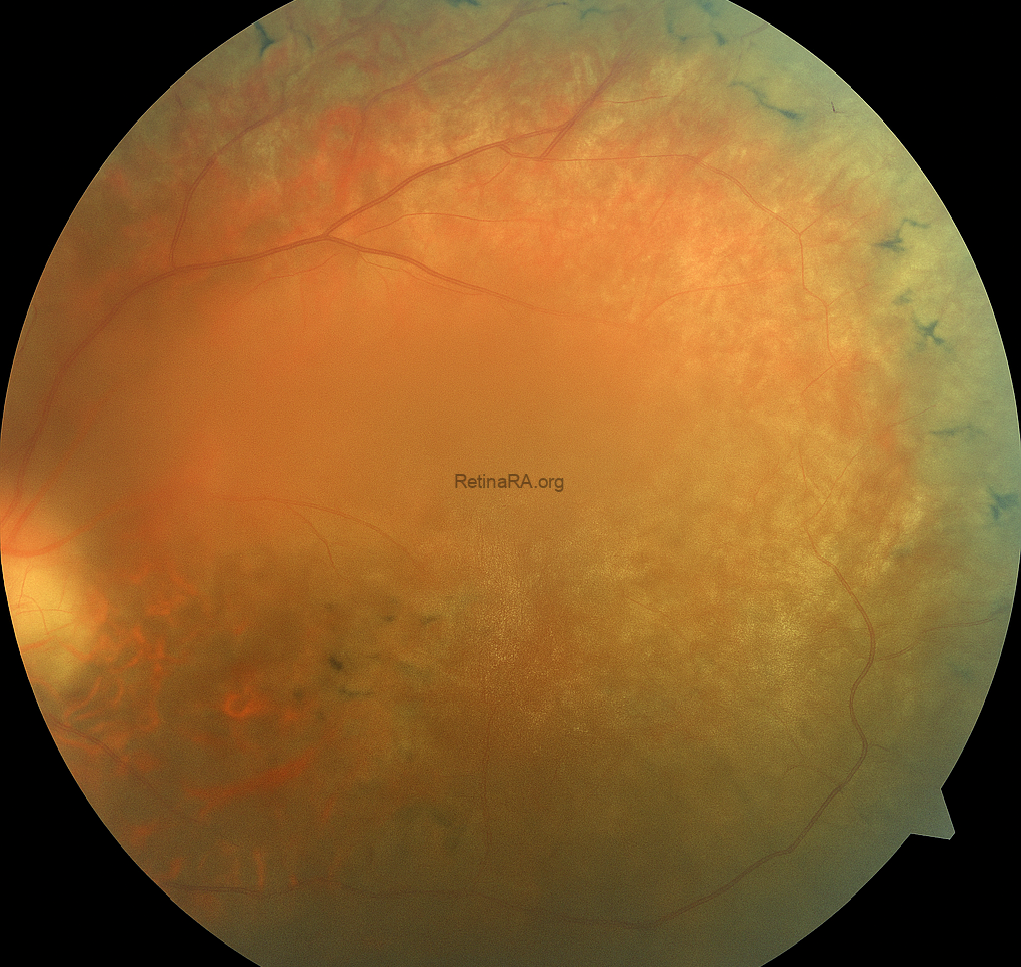
In Color fundus photography, in the right eye, attenuated retinal vessels, waxy pallor of the optic disc, and prominent bone spicule-like pigmentary deposits are observed in the mid-peripheral retina, consistent with rod-cone dystrophy. The overall image quality is sufficient to appreciate these hallmark findings. In contrast, the left eye fundus appears significantly blurred, limiting the detailed visualization of retinal structures. This reduced image clarity is attributed to media opacity secondary to cataract formation, which is a common ocular comorbidity in patients with syndromic retinal degenerations.
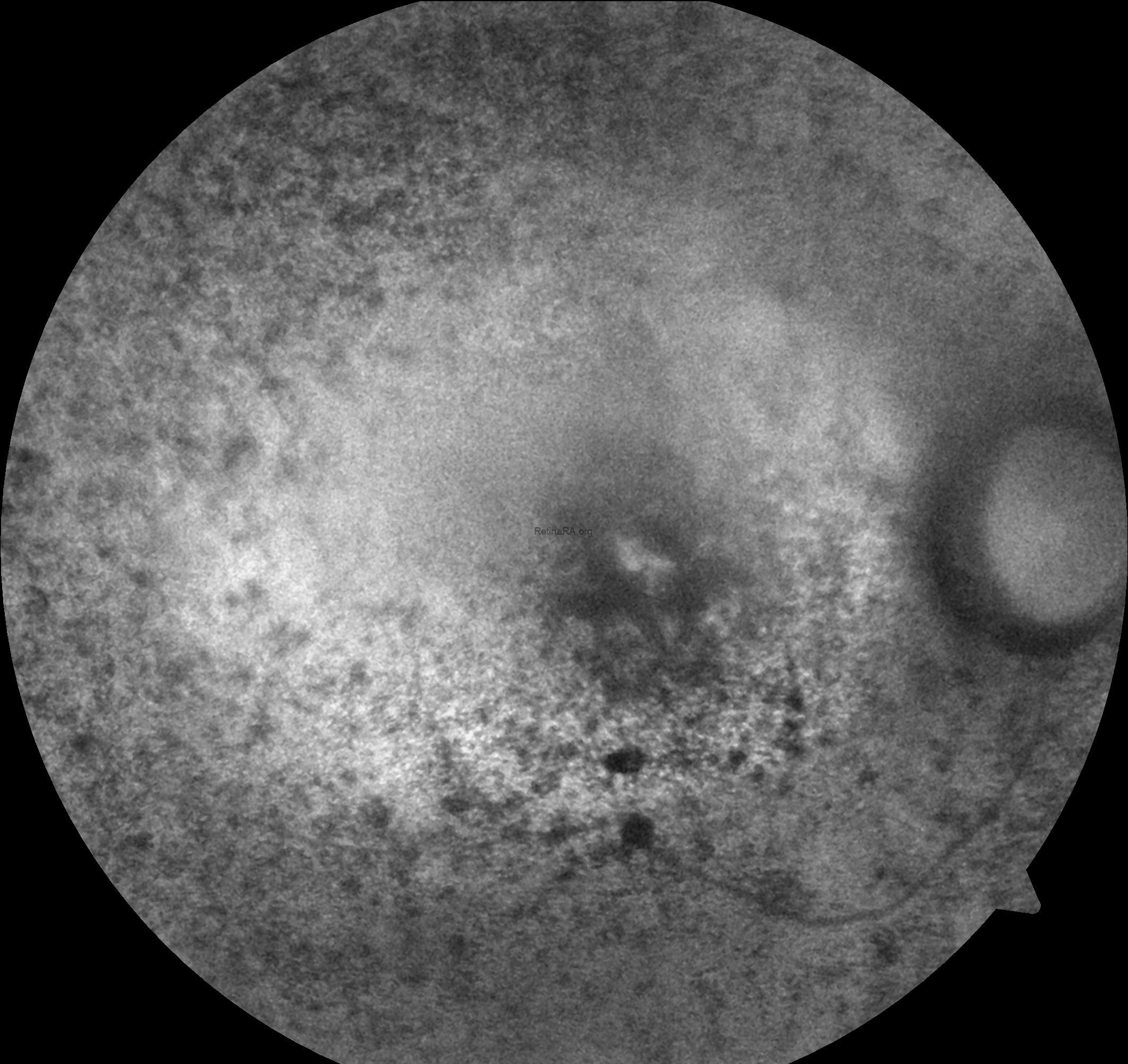
Fundus autofluorescence imaging of the right eye demonstrates a diffuse hypoautofluorescent pattern throughout the mid-peripheral retina, corresponding to areas of photoreceptor and retinal pigment epithelium (RPE) loss. A relatively preserved central autofluorescence signal suggests partial macular sparing, which is commonly observed in early to mid-stage rod-cone dystrophies. These findings are in line with the progressive retinal degeneration seen in Bardet-Biedl syndrome. However, FAF imaging of the left eye could not be obtained due to significant lens opacification secondary to cataract, which hindered adequate visualization of the posterior pole.
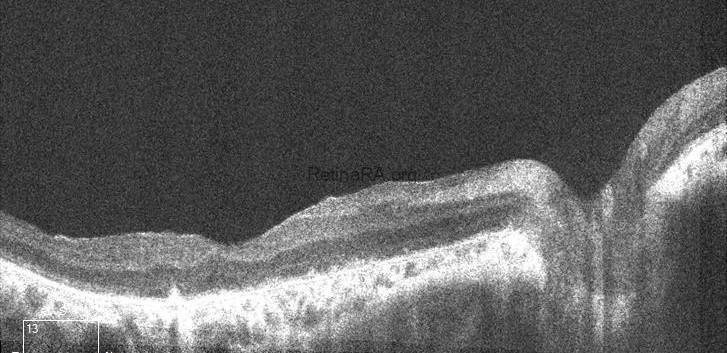
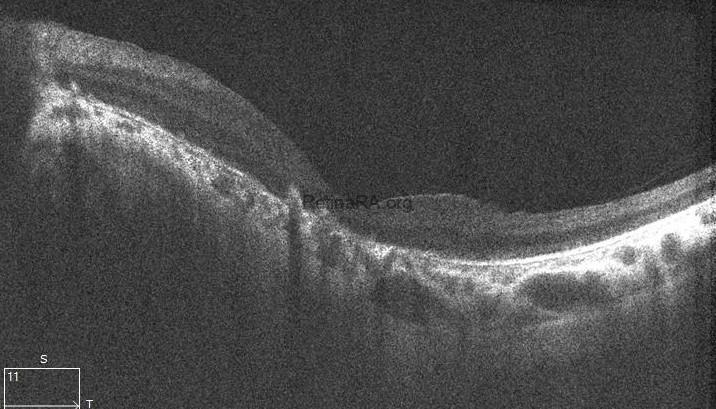
The scan of the right eye shows marked thinning of the outer retinal layers, particularly involving the outer nuclear layer and ellipsoid zone. The foveal contour is preserved; however, there is significant disruption and attenuation of the IS/OS junction and RPE band, consistent with advanced rod-cone dystrophy. In the left eye, generalized outer retinal atrophy is seen with loss of the photoreceptor integrity line and irregularity in the RPE layer. The foveal depression is shallow, and parafoveal architecture is disorganized, indicating chronic degenerative changes.
Bardet-Biedl syndrome (BBS) is a rare autosomal recessive disorder classified among the ciliopathies. It is characterized by a constellation of systemic manifestations including retinal dystrophy, polydactyly, obesity, renal abnormalities, and cognitive impairment. Retinal degeneration is often the earliest clinical feature, typically presenting as a rod-cone dystrophy with progressive vision loss beginning in childhood. The retinal phenotype observed in BBS closely resembles retinitis pigmentosa and is often regarded as a syndromic form of this condition, sharing characteristic features such as peripheral pigmentary changes, night blindness, and visual field constriction. The underlying pathology involves defective ciliary function affecting photoreceptor maintenance and signaling, ultimately leading to gradual photoreceptor cell loss.
Multimodal imaging in Bardet-Biedl syndrome reveals characteristic features of retinal degeneration. Fundus photography typically shows attenuated vessels, waxy optic disc pallor, and bone spicule pigmentation in the mid-periphery. Fundus autofluorescence often demonstrates patchy areas of hypoautofluorescence correlating with RPE loss. OCT reveals outer retinal thinning, disruption of the ellipsoid zone, and foveal architecture loss in later stages. Electroretinography shows diminished or extinguished scotopic and photopic responses, confirming widespread rod-cone dysfunction.
Currently, there is no curative treatment for Bardet-Biedl syndrome, and management is primarily supportive and multidisciplinary. Ophthalmologic follow-up focuses on maximizing residual vision through low vision aids, environmental adaptations, and treatment of associated conditions such as cataract or refractive errors. Given the progressive nature of the retinal degeneration, genetic counseling is crucial for affected families.
Credit: M. Giray Ersoz, MD, FEBO
Biruni University School of Medicine, Department of Ophthalmology, Istanbul, Turkey
Instagram accounts: @retina.review and @retina.dr.girayersoz
and Sepideh Lotfi, MD
Biruni University School of Medicine, Department of Ophthalmology, Istanbul, Turkey
Instagram accounts: @sepidls
