Alport syndrome is an inherited glomerular basement membrane disease caused by mutations in the COL4A3/4/5 genes. The most common inheritance is X-linked dominant, followed by autosomal recessive and autosomal dominant. Cases with Alport syndrome may develop hematuria, proteinuria, renal failure (edema and hypertension), hearing loss, and ocular abnormalities affecting the cornea, lens, and retina.
Characteristic ocular features of Alport syndrome are corneal opacities, anterior lenticonus and cataract, central perimacular and peripheral coalescing fleck retinopathies, and temporal retinal thinning. Rarely, posterior polymorphous corneal dystrophy, a macular hole, or a maculopathy impairs vision. Lenticonus, corneal dystrophy, central and peripheral fleck retinopathies, temporal retinal thinning, and giant macular hole are all highly suspicious for the diagnosis of Alport syndrome.
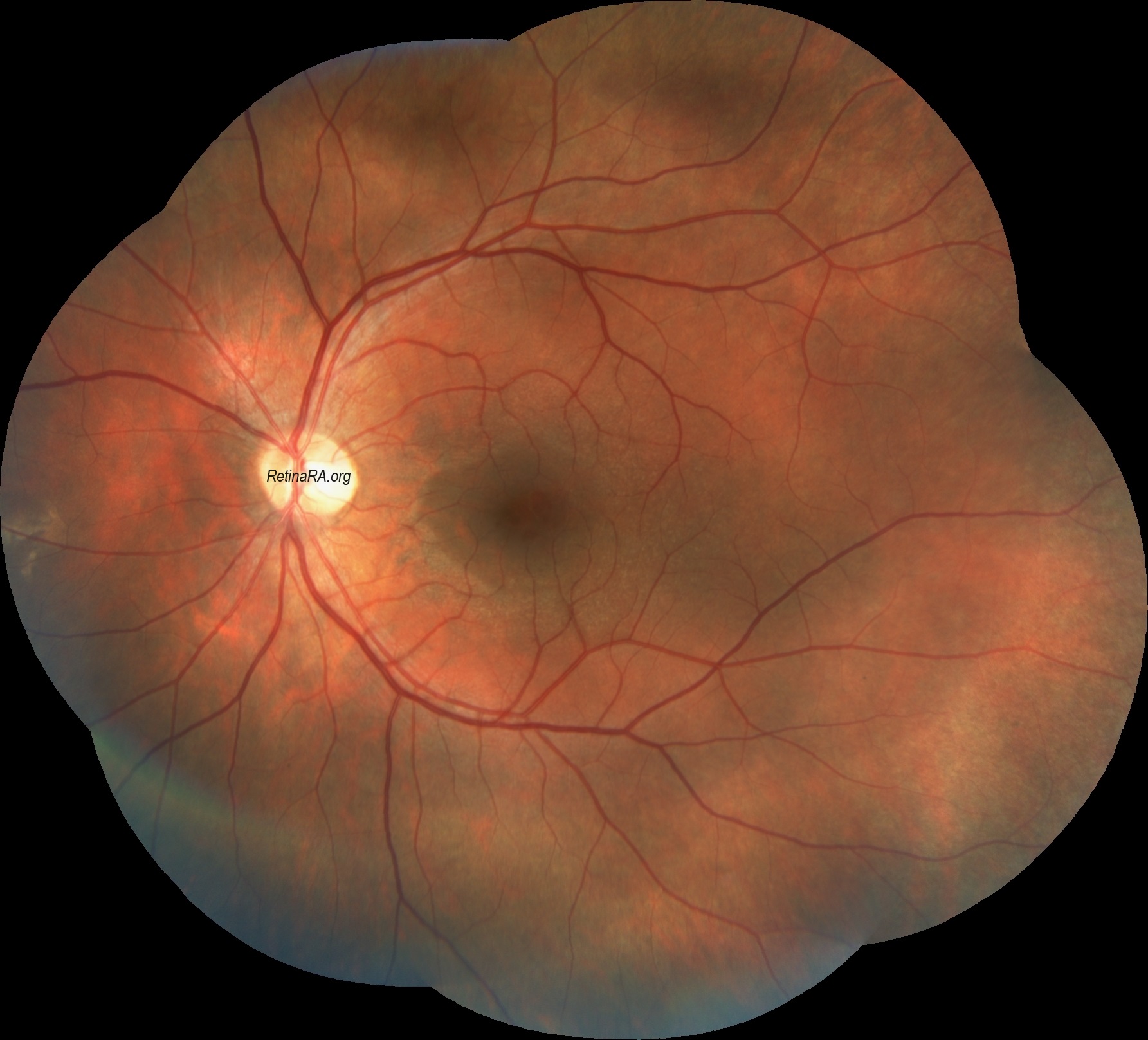
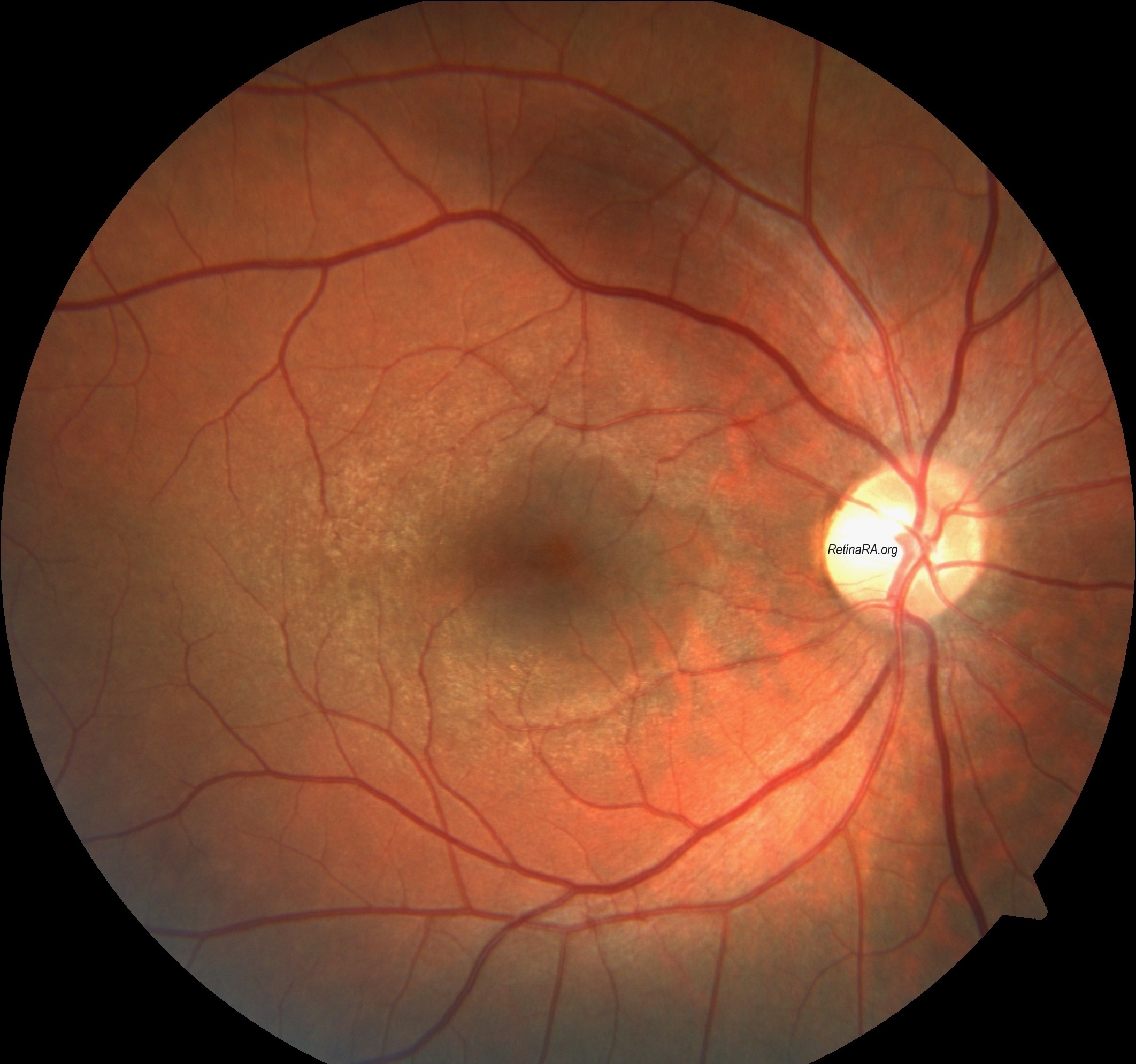
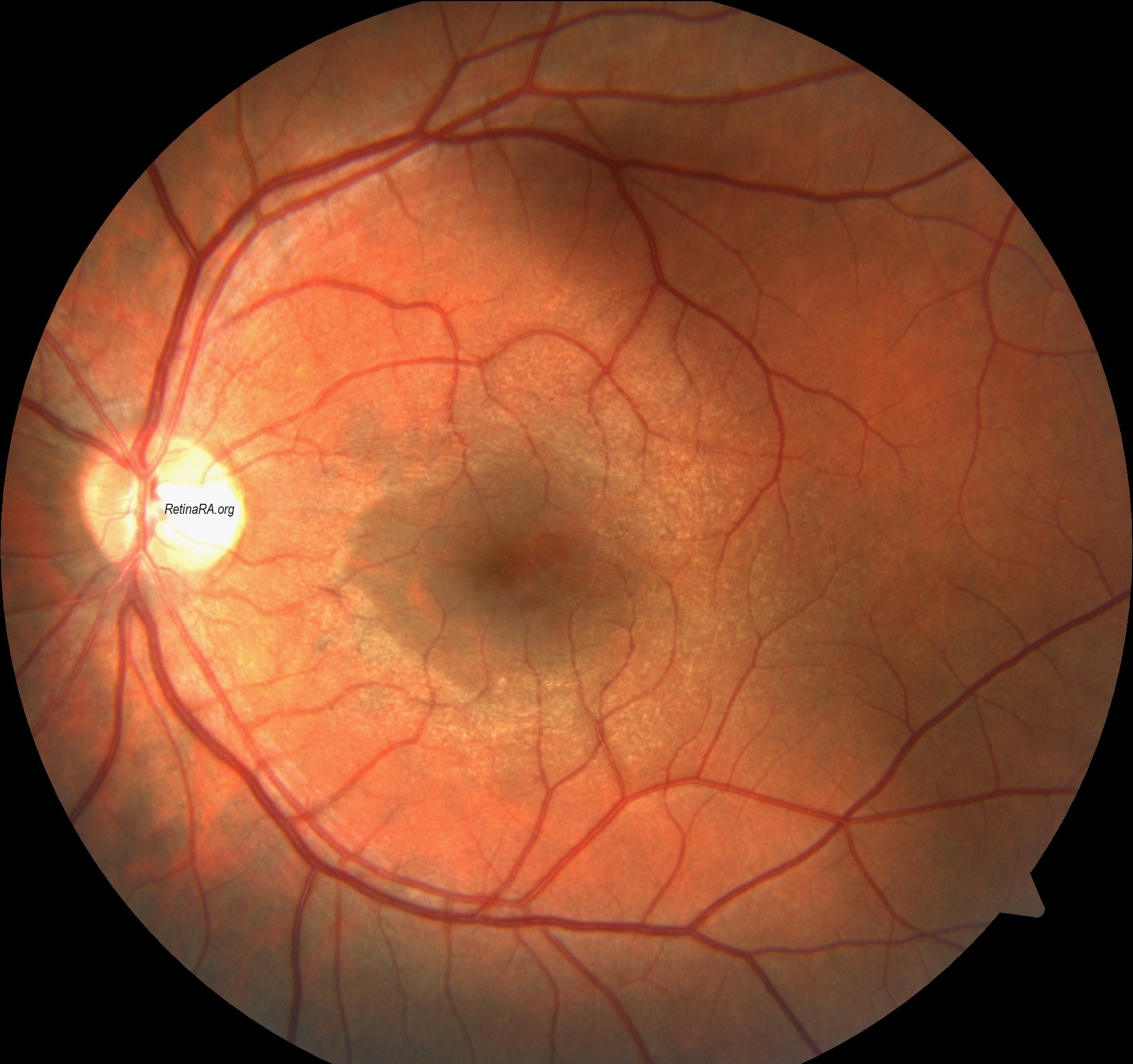
Temporal retinal thinning is very common in men and women with X-linked Alport syndrome, and with recessive disease. The lozenge, dull macular reflex, foveopathy, and lamellar and macular holes all affect the temporal retina and reflect retinal thinning of both the inner limiting membrane and Bruch’s membrane. Temporal retinal thinning is apparent on retinal color photographs as the dull macular reflex or lozenge with a larger, more oval shape rather than the normal round foveal reflex. Thinning is confirmed with retinal thickness measurements in the <5th percentile on OCT. Although thinning is common in all forms of Alport syndrome and is less diagnostically sensitive than peripheral retinopathy, its demonstration is more objective. Thinning occurs with retinal ischemia, but this is not the case in non-Alport renal failure. Retinal function tests are normal only when there is thinning. Vision is not affected.
Credit: Kemal Tekin, M.D., from Ulucanlar Eye Training and Research Hospital
Instagram accounts: @retina.academy and @dr.kemaltekin